(英文)
Re-analysis of a retracted paper: Detection of chromosomal aberrations
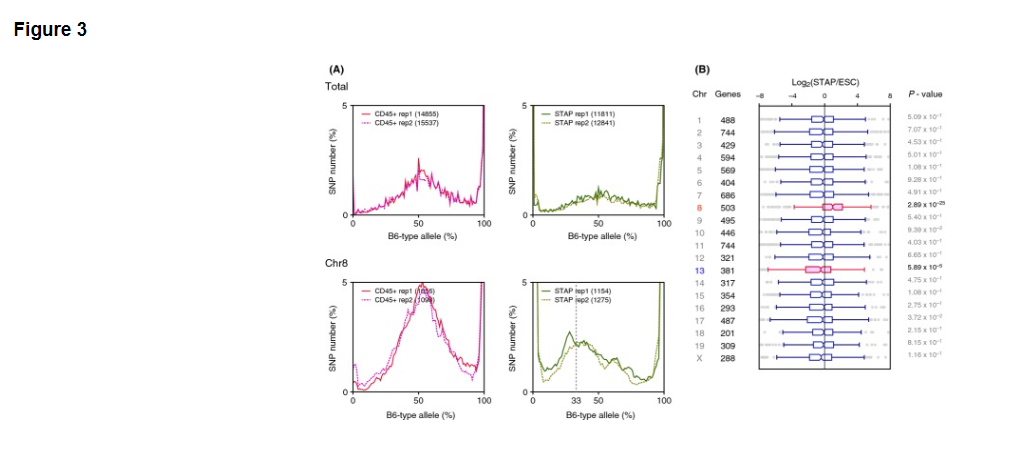
SNP distribution analysis can also be applied to detect aneuploidy. In examining allele frequencies for each chromosome, abnormal chromosomes are assumed to have skewed distributions when paired chromosomes do not have the equal number of duplicates. As shown in Fig. 3A, chromosomal analysis showed abnormality of chromosome 8 in the STAP cells used in the original study. We can expect the peak allele frequency to occur at approximately 50% if a cell has two chromosomes from parents of different strains. The peak allele frequency of the STAP cells, however, was at approximately 33%, meaning that one of the two chromosomes appears to be duplicated leading to three copies of chromosome 8.
撤回された論文の再分析:染色体異常の検出
SNPの分布分析は異数性を検出するためにも適用することができる。各染色体についての対立遺伝子頻度を調べる際には、対の染色体が同数の複写をもたないときに、異常な染色体が歪んだ分布を持つのだと推定される。図3Aに示すように、染色体分析は最初の研究で使われたSTAP細胞における8番染色体の異常を示している。細胞が異なる株の親からの2つの染色体を持っている場合には、我々はピーク対立遺伝子頻度が約50%に起こることを期待することができ.る。 STAP細胞のピークの対立遺伝子頻度は、しかし、約33%であった。それは2つの染色体の一つが染色体8の3つのコピーをもたらすように複製されたように見える。
(英文)
The STAP cells used in the experiments were derived from 129 and B6 cells, and the duplicated chromosome is presumed to be from the 129 parent because it contained only non-B6 SNP alleles. It is notable that trisomy 8 is the most common chromosomal aberration in mouse ESCs, with 31 of 97 examined cell lines reported to carry this abnormality (Mayshar et al. 2010). ESCs having trisomy 8 have a growth advantage, but chimeras will not transmit the mutation to the germ-line (Ben-David et al. 2013), and trisomy 8 in mice results in prenatal death by day 12 or 13 (Kim et al. 2013).
実験で使用されたSTAP細胞は129とB6細胞由来である。重複染色体は129の親からのものであると推定される。なぜならそれが唯一の非B6のSNP対立遺伝子を含んでいたためである。トリソミー8がマウスES細胞の中での最も一般的な染色体異常であることは注目に値する。それは97回の細胞株を調べたうちの31回この異常をもつと報告されていることである(Maysharら、2010)。トリソミー8を持つES細胞は生育が優性であるが、キメラは生殖系列への変異を引き起こさず(Ben-David et al. 2013)、マウス内のトリソミー8は12日目または13日目での出生前死亡をもたらします (Kim et al. 2013)。
(Fig. 3のリジェンド)
Figure 3. Trisomy detected by SNP analysis of RNA-seq data. (A) Allele frequency distributions of whole chromosomes and chromosome 8. Only chromosome 8 of STAP cells had a peak that was not centered approximately 50%, indicating that chromosome 8 originating from the 129 strain was duplicated to produce trisomy of the chromosome. Unlike the RNA-seq data analyzed in Figs 1 and 2, the RNA-seq data analyzed in this figure were generated using the SMARTer reagent kit. (B) Expression analysis by chromosome. Only genes on chromosome 8 were significantly more expressed, and genes on chromosome 13 were significantly less expressed. P-values were calculated using two-sided Student t-tests.
[図3] RNA-seqデータのSNP解析によって検出されたトリソミー。 (A)全染色体と8番染色体の対立遺伝子頻度分布。STAP細胞の8番染色体のみが約50%を中心としないピークを持っていた。それは129株起源の8番染色体が染色体のトリソミー生成するように複製されたことを示す。図1及び図2で分析されたRNA-seqデータとは異なり、この図において分析されているRNA-seqデータはSMARTer試薬キットを使用して作られている。(B)染色体ごとの発現解析。 8番染色体上の遺伝子のみが有意に多く発現しし、13番染色体上の遺伝子が有意に低い発現している。 P値は二群スチューデントt検定を用いて計算されている。
(本文続き)
Because aneuploidy detection using transcriptome analysis has been reported (Gropp 1982; Liu et al. 1997), the expression of genes on each chromosome was analyzed in this study. Genes on chromosome 8 and chromosome 13 had significantly different expression patterns between STAP cells and ESCs. Chromosome 8 gene expression was 1.3 times higher in STAP cells than in ESCs (P-value = 2.89 × 10-25; Fig. 3B). This result is concordant with trisomy of chromosome 8 as detected by SNP analysis. The SNP allele frequency method used here can, therefore, be used to detect aneuploidy if we know the SNP genotype of the cells and if the control cells have a normal karyotype.
メッセンジャーRNA解析を用いた異数性検出が報告されているので(Gropp 1982; Liu et al. 1997)、各染色体上の遺伝子発現を本研究で分析した。 8番染色体と13番染色体上の遺伝子はSTAP細胞とES細胞の間で有意に異なった発現パターンを持っていた。第8染色体遺伝子発現はES細胞よりSTAP細胞において1.3倍高かった(P-value = 2.89 × 10-25; 図3B)。この結果はSNP解析によって検出された8番染色体のトリソミーと一致している。ここで使用されているSNP対立遺伝子頻度法は、したがって、我々が細胞のSNPの遺伝子型を知っていて、かつ、対照細胞が正常な核型を持っている場合には異数性を検出するために使える。
(英文)
Availability of SNP allele frequencies
The SNP allele frequency method detected contaminating cells in samples used to generate RNA-seq data, but the sensitivity of this method is dependent on the depth of sequence reads. Based on the limited number of RNA-seq datasets here, the graphs become too noisy to detect differences in the distributions if the number of available SNPs is fewer than approximately one thousand. For example, MEF cells having only 5948 SNPs generates a more noticeable amount of spikes than data for ESCs with 23 838 SNPs. SNPs assigned to each chromosome also suggested that the distributions could be very noisy when the number of SNPs become <1000 (Fig. S2 in Supporting Information). Cover ratios are also important for the sensitivity because the variance of binomial distribution depends on the number of trials. Therefore, when the average cover ratio is required to be 20× for all genes, the required read count will be approximately 1.3 × 109 nucleotides, roughly corresponding to a read count of approximately 1.1 × 109 bases, which was used to derive the distribution for MEFs (Fig. 1B).
SNP対立遺伝子頻度の有効性
SNP対立遺伝子頻度手法はRNA-seqデータを作成しようとするときに通常使用されるサンプル中の汚染細胞を検出するが、この手法の感度は配列リードの長さに依存する。ここで限られた数のRNA-seqデータセットを仮定すると、利用可能なSNPの数がおよそ千よりも少ない場合は、グラフは分配の違いを検出するにはあまりに雑音が多くなる。例えば、5948個のSNPを持つMEF<マウス胎児線維芽細胞>は23838個のSNPを持つES細胞のデータよりもスパイクのより顕著な量を生成する。各染色体に割り当てられたSNPはまた、SNPの数が1000より小さいときの分布が非常にノイズが多くなりがちなことを示唆している(サポート情報の図S2)。二項分布の分散は試行回数に依存しているため、感度にとってはカバー率も重要である。それ故、平均カバー率がすべての遺伝子の20倍であることが要求されている場合、必要な読み出し回数は、MEF<マウス胎児線維芽細胞>の分布を導出するために使用された約1.1×109塩基のリード回数にほぼ対応するところの、約1.3×109ヌクレオチドであろう(図1B)。
(英文)
Figure 1B also indicates some interesting aspects of genome stability of induced pluripotent stem (iPS) cells. The iPS cells generated from 129B6F1 had more homozygous SNPs of B6-type alleles. Although, as noted earlier, this may be the result of cellular contamination or this could be the result of differences in the properties of the cells used in the two experiments, there is also the intriguing possibility that the experimental process induced a transition of genotypes. As iPS cell engineering has been reported to induce genomic and/or epigenomic instability (Hussein et al. 2011; Chang et al. 2014), it will be important to examine allele frequencies of iPS cells in future studies.
図1Bはまた人工多能性幹細胞(iPS)のゲノム安定性のいくつかの興味深い側面を示している。 129B6F1から生成されたiPS細胞はB6型対立遺伝子のよりホモ接合したSNPを有していた。先に述べたように、これは細胞の汚染の結果であるかもしれないし、この二つの実験で使用された細胞の性質の違いの結果である可能性もあるが、実験プロセスが遺伝子型の転移を誘導したという魅力的な可能性もある。 iPS細胞工学がゲノム的な、および/または、ゲノム外環境的な不安定性を誘導すると報告されているように (Hussein et al. 2011; Chang et al. 2014)、今後の研究においてiPS細胞の対立遺伝子頻度を調べることが重要となるでしょう。
(英文)
SNP frequency differences detected in RNA-seq or other NGS experiments are probably not more accurate than those detected by direct observation of karyotypes; however, the method described in this study can be used to detect chromosomal abnormalities even if the test cells are no longer available, and from both genotype (SNPs) and phenotype (gene expression), which is expected to provide more reliable evidence than genotype or phenotype alone.
RNA-seqや他のNGS実験で検出されたSNP頻度の違いは、おそらく核型を直接観察することによって検出されるものと比べると、正確ではありません。しかし、この研究で記載された方法は、試験細胞がもはや利用不可能である場合でも、遺伝子型か表現型のみによるよりもより信頼できる証拠を提供することが期待されている遺伝子型(SNP)と表現型(遺伝子発現)の両者から染色体異常を検出するために使用することができます。
(英文)
One advantage of the SNP allele frequency method of this study is its potential ability to detect and determine the parental origin of chromosomes duplications. The other advantage over virtual karyotyping (Ben-David et al. 2013) is that it does not require control cells without aneuploidy because simulated models expect the allele frequencies of diploid cells would have peaks approximately 50% (Fig. 1A).
この研究のSNP対立遺伝子頻度法の1つの利点は染色体複製の親の起源を検出しかつ決定する、その潜在能力にある。仮想染色体分析(Ben-David et al. 2013)を介したもうひとつの利点は、異数性のない対照細胞を必要としないことである。なぜなら、シミュレートされたモデルはそもそも二倍体細胞の対立遺伝子頻度がピークを約50%に持っているだろうと期待しているからである(図1A)。
- 2019/05/14(火) 10:07:42|
- 遠藤論文
-
-
| コメント:0
(英文)
Results and discussion
Mathematical model and simulation
Diploid cells have pairs of homologous chromosomes, and genes are expressed from the paternal and maternal chromosomes at roughly the same frequency, except in the case of imprinted genes and for genes on sex chromosomes (DeVeale et al. 2012; Lagarrigue et al. 2013). The frequency of expression of a sequence from a parent is expected by chance to follow a binomial distribution. Because bias caused by PCR amplification can affect this distribution, the influence of PCR bias was examined. When we consider nonimprinting genes having heterozygous SNPs, the allele frequency is expected to be approximately 50% for samples that consist of only one cell type, and the unbalanced representation of certain SNPs caused by contamination should appear as a shift in the distribution peaks.
結果と検討
数理モデルとシミュレーション
二倍体細胞は相同染色体のペアを持っています。遺伝子は、インプリント遺伝子の場合と性染色体上の遺伝子を除き、父方と母方の染色体からおよそ同一の頻度で提供されている(DeVeale et al. 2012; Lagarrigue et al. 2013)。片親からの配列の発現頻度は、二項分布に従う見込みによって期待されている。 PCR増幅に起因するバイアスがこの分布に影響を与える可能性があるため、PCRバイアスの影響を調べた。ヘテロ接合のSNPを有するノンインプリンティング遺伝子を考える際、対立遺伝子頻度は、ひとつの細胞型で構成されたサンプルの約50%であると予測され、かつ汚染によって引き起こされた特定のSNPのアンバランスな表現は分布ピークのシフトとして表われてくるはずです。
(英文)
A simulation was carried out to illustrate how contaminating cells affect the simple binomial distribution of allele frequencies. When the number of the reference allele (A) is nA and that of the alternative allele (a) is na, the chance of detection of the reference allele is nA/(nA + na). In RNA-seq experiments, bias originating from PCR should be considered. To simplify the model, the PCR bias was incorporated by assuming that the sequences containing A and a were amplified 2α and 2β times, respectively. If we obtain N fragments of the locus with RNA-seq, the probability of k reference allele sequences is calculated as follows:
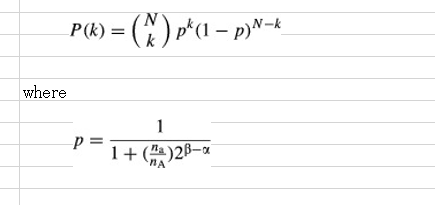
混入細胞が対立遺伝子頻度の単純な二項分布にどのように影響するかを図示するためにシミュレーションが実施された。参照対立遺伝子(A)の数をnA、代替対立遺伝子(a)のそれをnaとするとき、参照対立遺伝子の検出の可能性はnA /(nA+ na)である。 RNA-seqの実験では、PCRから生じるバイアスを考慮しなければならない。モデルを単純化するために、PCRバイアスはA及びaを含む配列がそれぞれ、2のα乗と2のβ乗倍に増幅されたと仮定して組み込まれた。もし我々がRNA-seqの持つ遺伝子座のN断片を得た場合は、k個の参照対立遺伝子配列の確率は次のように計算される。
(続き)
The simulation in Fig. 1A was carried out using conditions where N = 50 and β - α followed a Gaussian distribution, having standard deviation of 0 (no PCR bias) or 1 (high PCR bias). The simulation indicated that the variance of the distribution was highly dependent on PCR bias and that the mode of the distribution corresponded to the composition of SNP alleles. Allele frequencies of several sets of RNA-seq data from various cell types obtained from public databases were examined, and the results agreed with the simulation (Fig. 1B). Peaks at 0 and 100% might result from the homozygous SNPs in observed cells. An artificial contaminating situation was also generated with random sampling of RNA-seq datasets from two cell categories, pure C57BL/6 (B6) hematopoietic stem cells (HSCs), and a mixture of 129 and B6 embryonic stem cells (129B6F1 ESCs) at various ratios. The curve shape and peak positions varied along the ratio as shown in the mathematical simulation (Fig. 1C, gray line).
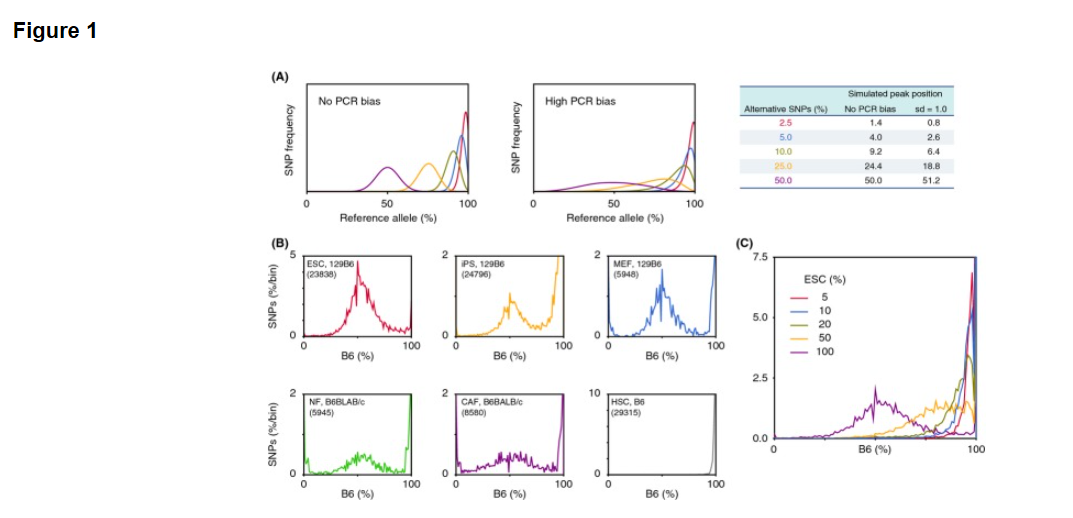
図1AのシミュレーションはN= 50で、かつ標準偏差が0(無PCRバイアス)または1(PCRバイアス)を有する ガウス分布に従ったβ-α条件を用いて行った。シミュレーションは分布の分散がPCRバイアスに依存的であったこと、分布様式が対立遺伝子の組成に対応していることを示した。公開データベースから得られた様々な細胞型からRNA-seqのデータのいくつかのセットの対立遺伝子頻度を調べた結果、シミュレーション(図1B)と一致した。 0%と100%でのピークは観察された細胞内のホモ接合のSNPに起因する可能性があります。人工汚染状況はまた2つのセルのカテゴリからのRNA-seqのデータセットのランダムサンプリングによって作られています。それは純粋なC57BL / 6(B6)の造血幹細胞(HSC)と各種129及びB6胚性幹細胞(129B6F1のESC)の各種比率の混合物の二つです。数学的シミュレーション(図1C、灰色の線)のように曲線形状とピーク位置はその比率に沿って変化しています。
(Fig. 1のリジェンド)
Figure 1. Allele frequency analysis of RNA-seq data. (A) Simulation of SNP allele frequencies using a modified binomial distribution. Peak position was determined by the composition of two alleles, and variance of the distribution was dependent on sd, standard deviation of simulated PCR bias. (B) SNP distributions in several cell types. ESCs (red, SRR1047502, 129B6F1 background), iPSs derived from fibroblasts (yellow, SRR1047504, 129B6F1), MEFs (blue, SRR104220, 129B6F1), normal fibroblasts (NFs; green, SRR1191170, B6 x BALB/c), cancer-associated fibroblasts (CAFs; purple, SRR1191171, B6 x BALB/c), and HSCs (gray, SRR892995, B6). The number of applied SNPs for each cell type is shown in parentheses in each box. (C) Allele frequency of HSC samples contaminated with different percentages of ESCs as shown.
[図1] RNA-seqデータの対立遺伝子頻度解析。(A)修正された二項分布を用いたSNPの対立遺伝子頻度のシミュレーション。ピーク位置は、2つの対立遺伝子組成物により決定され、分布の分散はsd、即ちシミュレートされたPCRバイアスの標準偏差に依存している。(B)いくつかの細胞型におけるSNP分布。ESCs<ES細胞>(赤、SRR1047502、129B6F1背景) 、線維芽細胞から誘導されたiPSs<iPS細胞>(黄色、SRR1047504、129B6F1)、MEF<マウス胎児線維芽細胞:フィーダー細胞>(青、SRR104220、129B6F1)、正常な線維芽細胞(NFs;緑、SRR1191170、B6 X BALB/ c )、癌化した線維芽細胞(CAFs;紫、SRR1191171、B6 x BALB / c)及びHSCs<造血幹細胞>(灰色、SRR892995、B6)。各細胞型のために適用されたSNPの数は各ボックス内の括弧内に示されている。 (C)示されたES細胞の異なる割合で汚染された造血幹細胞資料の対立遺伝子頻度。
(本文続き)
Re-analysis of STAP paper: Genotype analysis of fibroblast growth factor-induced stem cells (FI-SCs)
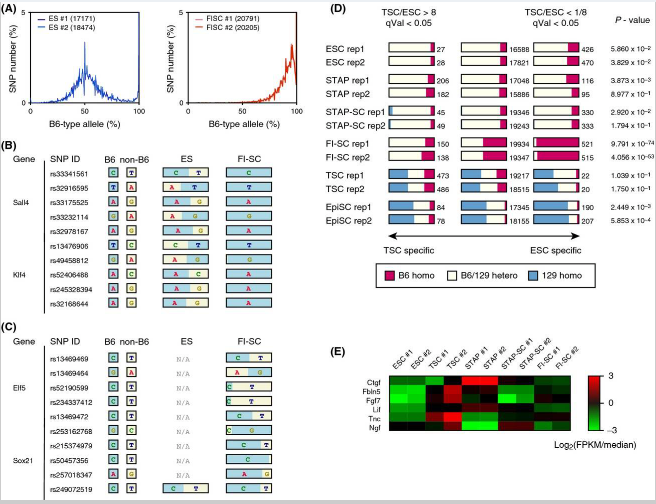
This study examined how SNP allele frequencies in RNA-seq data can be used to show properties of the dataset. Obokata et al. recently reported the phenomenon of STAP, the induced cellular reprogramming of committed somatic cells into pluripotent stem cells that can produce embryonic and placental tissues when injected into blastocysts (Obokata et al. 2014a,b). The allele frequency approach described above was used to examine the NGS dataset provided by the researchers. Allele frequencies between reference allele (equivalent to B6 genotype for dbSNP) and alternative allele (corresponding to 129 genotype in this study) were examined in RNA-seq data from seven replicate experiments obtained using the TruSeq reagent (Figs 2A and S1 in Supporting Information).
STAP論文の再解析: 線維芽細胞増殖因子によって誘導される幹細胞(FI-SC
)の遺伝子型解析
この研究はRNA-seqデータの中のSNP対立遺伝子頻度がいかにしてデータセットのプロパティを表示しうるに至るのかを検討している。 小保方らは最近STAP現象を報告した。それは胚盤胞に注入された場合に胚および胎盤組織を作り出すことができる多能性幹細胞へと変化した体細胞の誘導細胞再プログラミングを意味する(Obokata et al. 2014a,b)。上述の対立遺伝子頻度のアプローチは研究者らによって提供されているNGSデータセットを調べてきたものである。参照対立遺伝子(dbSNPのB6遺伝子型に相当)と、代替の対立遺伝子(この研究では129の遺伝子型に対応する)間の対立遺伝子頻度は、TruSeq試薬を使用して得られた7回の反復実験から得られたRNA-seqデータの中で検討されている(サポート情報の図2AおよびS1) 。
(英文)
The allele distributions of six of the seven experiments showed the equal representation of parental chromosomes expected in Fig. 1A. For the experiments that involved ESCs, STAP cells, and STAP stem cells (STAP-SCs), there were no 0% peaks (Fig. S1 in Supporting Information), possibly because the cells were obtained from mice backcrossed in the laboratory that may have a different genotype than those in the public database.
7回の実験中6回の対立遺伝子分布は図 1Aで期待されている親の染色体と同じ表現を示した。ES細胞、STAP細胞、およびSTAP幹細胞(STAP-SCS)を含む実験では全くの0%のピークは無かったが(サポート情報の図S1)、おそらく細胞が実験室で戻し交配されたため、公開データベース (J. Sharif and K. Isono, personal communication)のものとは異なる遺伝子型のマウスから得られたからであろう。
(Fig. 2のリジェンド)
Figure 2. SNPs detected in FI-SC mRNAs indicating contamination. (A) Allele distributions obtained from ESC and FI-SC RNA-seq experiments used in the STAP paper. Both ESCs (blue) and FI-SCs (red) are annotated as having a 129B6F1 genetic background. The number of applied SNPs for each experiment is shown in parentheses in the boxes. (B) SNPs detected in Sall4 and Klf4, which are highly expressed in ESCs. B6-type alleles are shown in blue and 129-type alleles (i.e., non-B6) are in yellow. (C) SNPs detected in the TSC-specific genes Elf5 and Sox21. (D) The number of homozygous/heterozygous SNPs observed in the stem cells used in the original paper. Only the composition observed in FI-SCs would be predicted to affect gene expression. P-values were calculated using Fisher's exact test of genotype distribution between TSC-specific genes and ESC-specific genes. Rep1 and rep2 denote two replicated experiments. (E) Heatmap of representative cytokine and extracellular matrix genes that are highly expressed in MEFs. Normalized log ratios of fragments per kilobase of exon per million reads (FPKM) against the medians of all samples were shown.
[図2] 汚染を示すFI幹細胞のmRNAで検出されたSNP。 (A)STAP論文に使用されたES幹細胞とFI幹細胞のRNA-seqの実験から得られた対立遺伝子分布。ES幹細胞(青)とFI幹細胞(赤)の両方とも129B6F1遺伝子背景を有していると注釈されている。各実験のために適用されたSNPの数は、ボックス内の括弧で示されている。(B)ES幹細胞で高頻度で発現されるSall4及びKlf4で検出されたSNP。 B6型対立遺伝子は青で、129型対立遺伝子(すなわち、非B6)は黄色で示されている。 (C)TS細胞特異遺伝子Elf5及びSox21で検出されたSNP。 (D)元の論文で使用された幹細胞で観察された沢山のホモ接合/ヘテロ接合SNP。 FI肝細胞の中で観察された組成物だけが遺伝子発現に影響を与えると予測される。 P値はTS細胞特異遺伝子およびES細胞特異遺伝子間の遺伝子型分布のフィッシャーの正確確率検定を用いて計算されている。 REP1およびREP2は、2つの反復実験を表す。(E)代表的なサイトカインおよび高頻度で胎児線維芽細胞に発現る細胞外マトリックス遺伝子のヒートマップ。全サンプルの中央値に対する万単位読み取り断片あたりの千単位エクソン断片の正規ログ比(FPKM)が示されている。
(本文続き)
Surprisingly, FI-SCs that were annotated as coming from the F1 129Sv (129) and B6 cell populations did not show the allele distribution pattern of unbiased nonimprinting genes (Fig. S1 in Supporting Information). The distribution was more similar to that of cells with unequal chromosomes. These FI-SCs were reported to be induced from STAP cells with Fgf4 and to have characteristics similar to trophoblast stem cells (TSCs), such as their gene expression profiles and potential to contribute to the placenta (Obokata et al. 2014a).
驚くべきことにF1 129SV(129)とB6の細胞集団由来と注釈されているFI幹細胞はバイアスのないノンインプリンティング遺伝子の対立遺伝子分布パターンを示さなかった(サポート情報の図S1)。分布は不均等な染色体を有する細胞のものにより類似している。これらのFI肝細胞はFGF4<線維芽細胞増殖因子-4 >によったSTAP細胞から誘導され、かつそれらの遺伝子発現の特徴と胎盤に貢献する能力のように、栄養膜細胞(TS細胞)に似た特性を有することが報告されている(Obokata et al. 2014a)。
(英文)
The obvious difference in the FI-SC curve from the 129B6F1 genotype, combined with the fact that the majority of SNPs were similar to B6, suggested that the FI-SCs originated from neonatal mice of a nearly pure B6 background. Further analysis of gene expression patterns suggested that the heterogeneity of SNPs between B6-type allele and non-B6 could be caused by the expression characteristics of genes. As shown in Fig. 2B, SNPs expected to be heterogeneous between 129 (i.e., non-B6) and B6 were examined in several ESC marker genes. ESCs carried alleles from both the 129 and B6 backgrounds at these loci, but the FI-SCs, although described as having the same genetic background as the ESCs (Obokata et al. 2014a), carried only SNPs from B6. This dominance of the B6 genotype was not observed in TSC marker genes (Fig. 2C).
大多数のSNPがB6と似ているという事実と組み合わせると、129B6F1遺伝子型とFI幹細胞曲線の明らかな差異はFI幹細胞がほぼ純粋なB6バックグラウンドの新生仔マウスに由来することを示唆している。遺伝子発現パターンの更なる分析は、B6型対立遺伝子と非B6間のSNPの不均一性が遺伝子発現特性に起因することが示唆されている。図2Bに示めされているように、129(すなわち、非B6)およびB6間で異質であることが期待されているSNPは、いくつかのES細胞マーカー遺伝子の中で調べられている。ES細胞はこの遺伝子座において129とB6の両方のバックグラウンドからの対立遺伝子を持ち込んでいるが、FI幹細胞は、ES細胞と同じ遺伝子背景を持つと書かれているにもかかわらず(Obokata et al. 2014a)、B6からの対立遺伝子しか持ち込んでいない。 B6遺伝子のこの優勢はTS細胞のマーカー遺伝子には観察されなかった(図2C)。
(英文)
The FI-SC specificity was not limited to the genes shown in Fig. 2B and C. When all heterogeneous SNPs were classified into three groups, SNPs in ESC-specific genes, SNPs in TSC-specific genes, and SNPs in other genes, only FI-SCs had widely heterozygous SNPs for these groups (Fig. 2D). If all included cells in a sample share the same cellular features, one would not expect to see this phenomenon of particular gene sets having different genotypes.
FI幹細胞の特異性は図2B及びCに示す遺伝子に限定されなかった。すべての異質SNPが、ES細胞に特異的遺伝子のSNP、TS細胞に特異的遺伝子のSNP、および他の遺伝子のSNPの3つのグループに分類されているとき、FI幹細胞のみがこれらのグループに広くヘテロ接合のSNPを有していた(図2D)。試料中に含まれるセルのすべが同じ細胞の特徴を共有している場合、異なる遺伝子型を有する特定の遺伝子セットのこの現象は見られ得ないであろう。
(英文)
Because the FI-SCs showed a specific genotype at some TSC markers, they may have been contaminated with TSCs. Feeder cells, however, could be another source of contamination, as the FI-SCs were cultured with mouse embryonic fibroblast (MEF) feeder cells whose genotype was not described in the original paper. For this study, the expression of marker genes for MEFs was examined and compared with the expression of ESC and TSC markers, and the results indicated the absence of expression of these MEF genes in FI-SCs (Fig. 2E). The probability of contamination by MEFs is therefore negligible, and the most likely explanation for the skewed distribution of allele frequencies detected in the duplicated RNA-seq experiments is that the FI-SC population originated from two cell types: ESC-like cells having a B6 genetic background and TSC-like cells having a genotype similar to that of CD1, which is a mouse strain other than B6 and 129.
FI幹細胞はいくつかのTS細胞マーカーで特定の遺伝子型を示したので、それらはのTS細胞で混入汚染されている可能性がある。しかしながら、 FI幹細胞がその遺伝子型が元の論文に記載されていないマウス胚性線維芽(MEF)フィーダー細胞とともに培養されていたとしたら、フィーダー細胞も混入汚染の他の原因でありうる。この研究にとって、MEF<マウス胚性線維芽細胞>のためのマーカー遺伝子の発現は調べられており、かつ、ES細胞とTS細胞のマーカー発現と比較されていて、その結果はFI幹細胞の中のこれらのMEF遺伝子の発現の欠如を示している(図2E)。 従ってMEF<胚性線維芽細胞>の混入の可能性は無視でき、かつ、重複RNA-seqの実験で検出された対立遺伝子頻度の傾斜分布の最も可能性の高い説明は、FI幹細胞の集団が次の2つの細胞型に由来していることである:B6遺伝子背景を有するES様細胞と、B6と129以外のマウス株で、CD1と同様の遺伝子型を有するTS様細胞。
- 2019/05/14(火) 10:01:28|
- 遠藤論文
-
-
| コメント:0
(英文)
Quality control method for RNA-seq using single nucleotide polymorphism allele frequency
Takaho A. Endo*
Article first published online: 21 SEP 2014
1. Top of page
2. Abstract
3. Introduction
4. Results and discussion
5. Conclusion
6. Experimental procedures
7. Acknowledgements
8. References
9. Supporting Information
『一塩基多型対立遺伝子頻度を用いたRNA-seqのための品質制御方法』
Takaho A. Endo*
Article first published online: 21 SEP 2014
1.ページ先頭
2.要約
3.導入
4.結果と検討
5.結論
6.実験経過
7.謝辞
8.照会
9.参考
(英文)
Abstract
RNA sequencing (RNA-seq) provides information not only about the level of expression of individual genes but also about genomic sequences of host cells. When we use transcriptome data with whole-genome single nucleotide polymorphism (SNP) variant information, the allele frequency can show the genetic composition of the cell population and/or chromosomal aberrations. Here, I show how SNPs in mRNAs can be used to evaluate RNA-seq experiments by focusing on RNA-seq data based on a recently retracted paper on stimulus-triggered acquisition of pluripotency (STAP) cells. The analysis indicated that different types of cells and chromosomal abnormalities might have been erroneously included in the dataset. This re-evaluation showed that observing allele frequencies could help in assessing the quality of samples during a study and with retrospective evaluation of experimental quality.
<概要>
RNAシークエンシング(RNA-seq)は個別遺伝子の発現レベルに関してのみならず、その細胞のゲノム配列についての情報をも提供する。我々が全ゲノム一塩基多型(SNP)変異体情報のあるメッセンジャーRNAデータを使用する場合、対立遺伝子頻度は細胞集団の遺伝子組成、および/または、染色体異常を示し得る。ここに私はどのようにしてmRNAの中のSNPが最近取り下げられた刺激惹起性多能性獲得(STAP)細胞論文に基づいたRNA-seqデータに焦点を当てることによって、RNA-seqの実験を評価するために使用し得るかという方法を示します。分析は異なった種類の細胞と染色体異常が誤ってデータセットに含まれているかもしれないことを示ました。この再評価は、対立遺伝子頻度の観察が研究中や実験の質の遡及評価をともなうサンプルの品質評価に役立つかもしれないことを示しています。
(英文)
Introduction
In molecular biological experiments, care must always be taken to prevent contamination from external sources, environmental substances, and undesired cells such as cocultured feeder cells. This has become increasingly important, as transcriptome analysis at the level of the single cell is now more common. Detecting contamination is often very difficult before sequencing experiments, and it is only when results are markedly different from predicted findings that researchers may suspect contamination. Various quality control methods have been proposed, but they focus mostly on other technical aspects of experiments (Wang et al. 2012). Here, I describe an approach to detect contaminating cells in studies using next-generation sequencing (NGS), particularly in transcriptome analyses based on RNA-seq techniques.
<導入>
分子生物学的実験では外部ソースや環境物質或いは共培養フィーダー細胞などの望ましくない細胞からの汚染を防ぐように常に注意が払われなければならない。このことは、単一細胞のレベルでのメッセンジャーRNA解析がより一般的となっている現在、ますます重要になっています。シークエンシング実験をせずに汚染を検出するのはしばしば非常に困難であり、また研究者が汚染を疑うのは結果が予期された発見と著しく異なった場合のみである。さまざまな品質管理手法が提案されているが、彼らは(Wang et al. 2012)実験の他の技術的な側面に主に焦点を当てている。ここでは私は次世代シーケンサー(NGS)を用いた研究において、とりわけRNA-seq技術に基づいたメッセンジャーRNA分析において、汚染細胞を検出するためのアプローチについて説明します。
(英文)
In RNA-seq experiments, NGS-derived mRNA sequences are compared with the target genome, and aligned reads are collected for all genes for which exon positions are provided in the database. One of the advantages of RNA-seq over microarray techniques is that it also provides some information about genomic sequences.
RNA-seqの実験においては、NGS由来のmRNA配列が標的ゲノムと比較される。そして、すべての遺伝子の整列させられた読み取り断片が集められる。またそのためにすべての遺伝子はエクソンの位置がデータベースに提供されている。DNAマイクロアレイ技術の中で、RNA-seqの利点の一つはゲノム配列に関するいくばくかの情報をも同時に提供することである。
(英文)
It is expected that mRNAs originate from both autosomal chromosomes and that the two (or more) alleles would thus be observed in the set of sequence fragments in NGS data. Skew in the frequency of alleles, as assessed by observed SNPs, can indicate several possible biological phenomena. The most preferable source of the skewed distribution, in terms of biological relevance, is allele-specific expression, such as genomic imprinting and mutation. However, another possible source of skewed allele frequencies is sample contamination if the contaminating cells have a different genomic background from genuine target cells.
mRNAが二つの常染色体に由来することと、それゆえ、二つ(またはそれ以上)の対立遺伝子がNGSデータの中のいくつかの系譜断片に観察されるかもしれないことは期待される。対立遺伝子頻度のゆがみは、観測されたのSNPによって評価されるように、いくつかの可能な生物学的現象を指示し得る。ゆがんだ分布の最も好ましい原因は、生物学的関係の条件下で、ゲノムインプリンティング、突然変異などの対立遺伝子の特異的発現が有った場合である。しかしながら、混入細胞が本物の標的細胞とは異なるゲノム背景を持っている場合には、歪んだ対立遺伝子頻度のもうひとつの可能な原因としてサンプルの汚染がある。
(英文)
Although allele frequencies are dependent on the PCR efficiency of each allele, and, in this analysis, variance of the distribution was especially dependent on PCR conditions, the average was approximately 50% for heterozygous SNPs, independent of the cell type.
対立遺伝子頻度は各対立遺伝子のPCR効率に依存しているけれども、またこの分析では分布の分散が特にPCR条件に依存していたが、細胞型とは独立して、平均はヘテロ接合のSNPの約50%であった。
(英文)
Allele frequency in RNA-seq has been used to detect imprinted genes (DeVeale et al. 2012; Lagarrigue et al. 2013). The approach described here extends the application of variation databases for the detection of contaminating cells in RNA-seq studies using heterozygous SNPs. Furthermore, this approach also provides a method for detecting chromosomal abnormalities. Skewed allele frequencies in a specific chromosome can be caused by aneuploidy. As aneuploidy can cause various types of abnormalities, we can exclude data from abnormal cells in studies when aneuploidy is not expected in the target tissue.
RNA-seqの中の対立遺伝子頻度はインプリント遺伝子を検出するために使用されてきている(DeVeale et al. 2012; Lagarrigue et al. 2013)。ここで説明するアプローチはヘテロ接合のSNPを用いたRNA-seqの研究の中で、汚染細胞汚検出のためのバリエーションデータベースの適用を拡張します。さらに、このアプローチは、染色体異常を検出する方法をも提供します。特定の染色体におけるゆがんだ対立遺伝子頻度は異数性によって引き起こされ得ます。異数性は多様なタイプの異常を引き起こしうるので、異数性が標的組織に期待されていない場合、我々は研究における異常細胞からのデータを除外することができる。
(英文)
This method is applicable for retrospectively evaluating the quality of experiments and is useful for interpreting results that are not apparently reproducible.
この方法は遡及的に実験品質を評価するために適用可能であると同時に、明らかに再現性がない結果を解釈するのに便利です。
- 2019/05/14(火) 09:48:44|
- 遠藤論文
-
-
| コメント:0
(英文)
Extended Data Figures
1. Extended Data Figure 1: Placental contribution of STAP cells. (295 KB)
a, Chimaeric mouse with STAP cells derived from CD45+ cells of B6GFP × 129/Sv mice (B6GFP, C57BL/6 line with cag-gfp transgene). Arrows indicate a placenta and a yolk sac. b, Cross-sections of yolk sac (top) and placenta (bottom). GFP-positive cells (arrows) were seen only in yolk sac and placenta of the STAP cell chimaera. Scale bars, 50 μm. c, Co-immunostaining showed that these GFP-positive cells (right) were found in the extra-embryonic endoderm-derived epithelial cells (pan-cytokeratin+ and overlying laminin+ basement membrane; left) of the yolk sac. Scale bar, 10 μm.
拡張データ図
1.拡張データ図1:STAP細胞の胎盤寄与。 (295 KB)
a、B6GFP×129 / Svマウス(B6GFP、cag-gfp導入遺伝子を有するC57BL / 6系統)のCD45 陽性細胞に由来するSTAP細胞のキメラマウス。 矢印は、胎盤および卵黄嚢を示す。 b、卵黄嚢(上)および胎盤(下)の断面。 GFP陽性細胞(矢印)は、STAP細胞キメラの卵黄嚢および胎盤においてのみ見られた。 スケールバー、50μm。 c、共染色結果は、これらのGFP陽性細胞(右)が卵黄嚢の胚外内胚葉由来上皮細胞(汎サイトケラチン陽性かつ重層ラミニン陽性の基底膜;左側)に見出されたことを示した。 スケールバー、10μm。
(英文)
2. Extended Data Figure 2: Trophoblast differentiation potential of Fgf4-induced stem cells. (749 KB)
a, b, Immunostaining (cross-section) of placentae obtained in the blastocyst injection assay with GFP (constitutive)-labelled ES cells (upper) or Fgf4-induced stem cells (bottom). Brown shows pan-cytokeratin and red shows GFP (ES cell or Fgf4-induced stem cell contribution). Regions indicated in a are shown in b. Fgf4-induced stem cells contributed to all layers of placentae, whereas no contribution was observed with ES cells. a, Scale bars, 5 mm. b, Scale bars, 50 μm. c, Pluripotent marker expression of Fgf4-induced stem cells. Scale bars, 50 μm.
d, e, Effects of Fgf4 withdrawal from Fgf4-induced stem cell culture. Unlike trophoblast stem cells (d, left), which generated multi-nucleated large cells (arrow) in the absence of Fgf4, Fgf4-induced stem cells (d, right) simply stopped proliferation and gradually died on Fgf4 withdrawal. Scale bars, 50 μm. This finding suggests that placental differentiation of Fgf4-induced stem cells in vivo may involve more than just Fgf4 signal suppression. e, The number of 4N and 8N cells increased within 6 days of Fgf4 withdrawal in trophoblast stem cells but not in Fgf4-induced stem cells.
2.拡張データ図2:Fgf4誘導幹細胞の栄養膜分化能。 (749 KB)
a,b,GFP(恒常性)標識ES細胞(上)またはFgf4誘導幹細胞(下)を用いた胚盤胞注入実験で得られた胎盤の免疫染色(断面)。 ブラウンは汎サイトケラチンを示し、赤はGFP(ES細胞またはFGF4誘導性幹細胞寄与)を示す。 aに示される領域は、bに示されている。 Fgf4誘導性幹細胞は胎盤のすべての層に寄与したが、ES細胞は寄与しなかった。 a、スケールバー、5mm。 b、スケールバー、50μm。 c,Fgf4誘導幹細胞の多能性マーカー発現。 スケールバー、50μm。
d,e,Fgf4誘導幹細胞培養からFgf4を除去した効果。 Fgf4の非存在下で大きな多能有核細胞(矢印)を形成する栄養膜幹細胞(d、左)とは異なり、Fgf4誘導性幹細胞(d、右)はFgf4を除去すると、単に増殖を停止し、徐々に死亡した。 スケールバー、50μm。 この知見は、インビボでのFgf4誘導幹細胞の胎盤分化は、単にFgf4シグナルの抑制以上のものを含む可能性があることを示唆している。 e、4Nおよび8N細胞の数は、栄養膜幹細胞においてFgf4の除去から6日間まで増加したが、Fgf4誘導性幹細胞では増加しなかった。
(英文)
3. Extended Data Figure 3: Transcriptome analyses of STAP cells shown by heat maps. (494 KB)
a, Heat maps of expression profiles of top-ranked up- and downregulated genes in STAP cells (Oct4-GFP+ clusters converted from CD45+ cells) compared to ES cells. Their respective expression levels in STAP stem cells, trophoblast stem cells and Fgf4-induced stem cells are shown. Absolute expression values are scaled by log2. The genes expressed differentially between ES cells and STAP cells tended to show more similar expression profiles to ES cells in STAP stem cells and Fgf4-induced stem cells than in trophoblast stem cells. Expression of some early endodermal lineage genes such as Gata4 and Sox17 was moderately elevated in STAP cells as compared to ES cells, whereas its biological significance remains elusive (these genes are shown to be strongly expressed in Oct4-GFP-dim cells1). b, Heat maps of expression profiles of top-ranked up- and downregulated genes in ES cells compared to CD45+ cells and their respective expression levels in STAP cells.
The genes expressed differentially between CD45+ and ES cells tended to show similar expression profiles in ES cells and STAP cells. c, Heat maps of expression profiles of representative genes implicated in haematopoietic lineage development in CD45+, ES and STAP cells. No strong correlation was seen between CD45+ cells and STAP cells in their expression profiles (a similar tendency of no correlation was seen for the data in b).
拡張データ図3:ヒートマップ<翻訳機訳注:個々の値のデータ行列を色として表現した可視化グラフ>によって示されるSTAP細胞のトランスクリプトーム<翻訳機訳注:特定の状況下において細胞中に存在する全てのmRNA(ないしは一次転写産物、 transcripts)の総体を指す呼称>解析。 (494 KB)
a,ES細胞と比較した、STAP細胞(CD45陽性細胞から変換されたOct4-GFP陽性クラスター)における上位ランクのアップ或いはダウンレギュレート<翻訳機訳注:転写物の増減により製造蛋白質の量が増減する>された遺伝子の発現プロファイルのヒートマップ。 STAP幹細胞、栄養膜幹細胞およびFgf4誘導幹細胞のそれぞれの発現レベルが示されている。 発現の絶対値はlog2(翻訳機訳注:Excel関数用語で2を底とする対数の意、数学定義の常用対数、自然対数のLog2ではない。)でスケールされている(翻訳機訳注:横の色分けバー)。ES細胞とSTAP細胞との間の遺伝子発現の違いは、STAP幹細胞と栄養膜幹細胞よりも、STAP幹細胞とFgf4誘導幹細胞に、より類似した発現プロファイルを示す傾向があった。 Gata4およびSox17のようないくつかの初期内胚葉系遺伝子の発現は、ES細胞と比較してSTAP細胞では適度に上昇したが、その生物学的意義は依然として分かっていない(これらの遺伝子はOct4-GFP弱発現細胞で強く発現することが示されている)。 b,CD45 陽性細胞と比較した、ES細胞におけるトップランクのアップ或いはダウンレギュレートされた遺伝子の発現プロファイルのヒートマップおよびSTAP細胞のそれらに対応する発現レベル。
CD45 陽性細胞とES細胞との間の発現遺伝子の違いはSTAP細胞においても同様の傾向があった。 c,CD45陽性、ES細胞およびSTAP細胞における造血系統発生に関与する代表的遺伝子の発現プロファイルのヒートマップ。 CD45陽性細胞とSTAP細胞との間には、その発現プロファイルにおいて強い相関は見られなかった(bのデータについても同様の傾向は見られなかった)。
(英文)
4. Extended Data Figure 4: Transcriptome analyses for genes implicated in cell-cycle control and induced pluripotent stem-cell conversion. (452 KB)
a, Comparison of expression values of genes involved in cell-cycle control in ES and STAP cells; the G to M cell cycle phases (upper), the cell cycle checkpoint and cell cycle arrest (middle), and the cell cycle regulation (bottom) are shown. Expression level was measured by log2 of mean normalized counts. b, Heat map for upregulated genes in cells undergoing reprogramming by ‘Yamanaka factors’[14]. c, Heat maps for upregulated genes in pre-iPS cells[15] (top) and in partially reprogrammed cells by Yamanaka factors (bottom)[14]. Expression level was measured by log2 of mean normalized counts. Differentially expressed genes were identified by the DESeq package[21] and only genes with a false discovery rate of 1% were selected for comparison, unless mentioned otherwise.
拡張データ図4:細胞周期制御および誘導された多能性幹細胞変換に関与する遺伝子のトランスクリプトーム解析。 (452 KB)
a,ES及びSTAP細胞における細胞周期制御に関与する遺伝子の発現値の比較; GからM細胞周期相(翻訳機訳注:細胞分裂のG0,G1,S,G2,Mの各段階)(上)、細胞周期チェックポイントおよび細胞周期停止段階(中)および細胞周期調節段階(下)が示されている。 発現レベルは平均正規化カウントのlog2によって測定されている。 b,「Yamanaka因子」[14]によって再プログラミングされている細胞におけるアップレギュレートされた遺伝子のヒートマップ。 c,pre-iPS細胞[15](上)およびYamanaka因子[14](下)による部分的に再プログラムされた細胞におけるアップレギュレートされた遺伝子のヒートマップ。 発現レベルは平均正規化カウントのlog2によって測定されている。。 示差的に発現された遺伝子は、DESeqパッケージ[21]によって同定され、他に言及されない限り、誤発見率1%の遺伝子のみが比較のために選択された。
(英文)
5. Extended Data Figure 5: Responses of Fgf4-induced stem cells to signal modifications. (333 KB)
a–f, JAK inhibitor treatment assay for Fgf4-induced stem cells. Fgf4-induced stem cells were cultured under feeder-free conditions and treated with 0.6 μM JAK inhibitor for 48 h. JAK inhibitor treatment assay eliminated ES cells (Oct4-GFP+) from the culture (a, b). The level of Oct4-GFP expression in Fgf4-induced stem cells, which was moderate, was maintained even after JAK inhibitor treatment (c, d; three independent experiments). Scale bar, 100 μm. e, f, For an additional control, Fgf4-induced stem cells were plated in trophoblast stem-cell medium containing Fgf4 together with Oct4-GFP ES cells that constitutively expressed BFP (the number of plated cells was one-tenth of that of plated Fgf4-induced stem cells). Whereas BFP-expressing colonies (ES-cell-derived) still expressed Oct4-GFP in trophoblast stem-cell culture medium after 2 days (e), no Oct4-GFP+ colonies from BFP-expressing ES cells were observed in the JAK-inhibitor-treated culture (f).
g, FACS analysis of integrin α7 expression in Fgf4-induced stem cells. Over 40% of Fgf4-induced stem cells strongly expressed both the pluripotency marker Oct4-GFP and the trophoblast marker integrin α7. The bottom panel shows an isotype control for integrin α7 antibody. In ES cells, integrin-α7-expressing cells were less than 0.1% (data not shown; three independent ES cell lines were examined).
拡張データ図5:Fgf4誘導幹細胞のシグナル修飾に対する反応。 (333 KB)
a-f,Fgf4誘導幹細胞に関するJAK阻害剤処理実験。 Fgf4誘導幹細胞をフィーダー細胞無しで培養し、0.6μMのJAK阻害剤で48時間処理した。 JAK阻害剤処理実験は、ES細胞(Oct4-GFP 陽性)を培養物(a、b)から除去した。 Fgf4誘導幹細胞におけるOct4-GFP発現のレベルは、中等度ではあるが、JAK阻害剤処理後でも(c、d; 3回の独立した実験)維持された。 スケールバー、100μm。 e,f, 加えて別のコントロール実験として、Fgf4誘導性幹細胞を、Oct4-GFPとともにBFPを恒常的に発現する ES細胞(播種細胞数はFgf4誘導幹細胞数の数の1/10)と共にFgf4を含有する栄養膜幹細胞培地に播種した。 BFPを発現するコロニー(ES細胞由来)が2日後に栄養膜幹細胞培養培地中でOct4-GFPを依然として発現した(e)のに対して、BFP発現ES細胞由来のOct4-GFP 陽性コロニーはJAK阻害剤添加培地では観察されなかった(f)。
g,Fgf4誘導幹細胞のインテグリンα7発現のFACS解析。 Fgf4誘導幹細胞の40%以上が多能性マーカーであるOct4-GFPおよび栄養膜マーカーであるインテグリンα7の両方を強力に発現した。 下のパネルは、インテグリンα7抗体のアイソタイプコントロールを示す。 ES細胞では、インテグリンα7発現細胞は0.1%未満であった(データは示していない; 3つの独立したES細胞株を調べた)。
(英文)
6. Extended Data Figure 6: Characterization of ES-like cells converted from Fgf4-induced stem cells and comparison of STAP cells with early embryos. (316 KB)
a, Immunohistochemistry of ES-like cells for trophoblast and pluripotency markers. ES-like cells converted from Fgf4-induced stem cells no longer expressed the trophoblast marker (integrin alpha 7), but they did express the pluripotency markers (Oct4, Nanog and SSEA-1). Scale bar, 100 μm. b, Pluripotency of ES-like cells converted from Fgf4-induced stem cells as shown by teratoma formation. Those cells successfully formed teratomas containing tissues from all three germ layers: neuroepithelium (left, arrow indicates), muscle tissue (middle, arrow indicates) and bronchial-like epithelium (right). Scale bar, 100 μm. c, MEK inhibitor treatment assay for Oct4-gfp Fgf4-induced stem cells in trophoblast stem-cell medium containing Fgf4. No substantial formation of Oct4-GFP+ colonies was observed from dissociated Fgf4-induced stem cells in MEK-inhibitor-containing medium. Scale bar, 100 μm.
d, Cluster tree diagram from hierarchical clustering of global expression profiles. Red, AU P values. As this analysis included morula and blastocyst embryos from which only small amounts of RNA could be obtained, we used pre-amplification with the SMARTer Ultra Low RNA kit for Illumina Sequencing (Clontech Laboratories). e, f, Volcano plot of the expression profile of STAP cells compared to the morula (e) and blastocyst (f). Genes showing greater than 10-fold change and P value 1.0 × 10(to the power of-6 )are highlighted in red and are considered up- (or down-) regulated in the STAP cells.
拡張データ図6:Fgf4誘導幹細胞から変換されたES様細胞の特徴付けおよびSTAP細胞と初期胚との比較。 (316 KB)
a,栄養膜マーカーおよび多能性マーカーに関するES様細胞の免疫組織化学。Fgf4誘導幹細胞から変換されたES様細胞は、栄養膜マーカー (integrin alpha 7)をもはや発現しなかったが、それらは多能性マーカー(Oct4、NanogおよびSSEA-1)を発現した。スケールバー、100μm。 b,テラトーマ形成によって示されたFgf4誘導幹細胞から変換されたES様細胞の多能性。これらの細胞は三胚葉すべてからの組織を含むテラトーマをきれいに形成した:神経上皮(左、矢印が示す)、筋肉組織(中、矢印が示す)および気管支様上皮(右)。 スケールバー、100μm。 c,Fgf4を含有する栄養膜幹細胞培地中のOct4-gfp陽性Fgf4誘導幹細胞に対するMEK阻害剤処理実験。 MEK阻害剤含有培地中の解離したFgf4誘導細胞から、Oct4-GFP陽性コロニーの実質的な形成は観察されなかった。 スケールバー、100μm。
d、グローバルな発現プロファイルの階層的クラスタリングによるクラスタツリー図。 赤、AU P値。 この分析には、少量のRNAしか得られない桑実胚および胚盤胞胚が含まれていたため、Illumina Sequencing(Clontech Laboratories)用のSMARTer Ultra Low RNAキットでプレ増幅を行った。 e,f,桑実胚(e)および胚盤胞(f)と比較したSTAP細胞の発現プロフィールの火山プロット。 10倍を超える変化およびP値1.0×10マイナス6乗を示す遺伝子は赤色で強調表示され、STAP細胞において上方(または下方)に調節されると考えられる。
<サプリテーブルは省略 >
- 2019/05/14(火) 09:33:49|
- レター論文
-
-
| コメント:0
(英文)
References
1. Obokata, H. et al. Stimulus-triggered fate conversion of somatic cells into pluripotency. Nature 505, 641–647 (2014)
2. Gurdon, J. B. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J. Embryol. Exp. Morphol. 10, 622–640 (1962)
3. Wakayama, T., Perry, A. C., Zuccotti, M., Johnson, K. R. & Yanagimachi, R. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 394, 369–374 (1998)
4. Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006)
5. Nagy, A., Rossant, J., Nagy, R., Abramow-Newerly, W. & Roder, J. C. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl Acad. Sci. USA 90, 8424–8428 (1993)
参考文献
1. Obokata, H. et al. 体細胞の多能性への刺激誘発性運命変換。 Nature 505, 641–647 (2014)
2. Gurdon, J. B. オタマジャクシの腸上皮細胞から採取した核の発生能。 J. Embryol. Exp. Morphol. 10, 622–640 (1962)
3. Wakayama, T., Perry, A. C., Zuccotti, M., Johnson, K. R. & Yanagimachi, R. 卵丘細胞核を注入した除核卵母細胞からのマウスの全期間発生。 Nature 394, 369–374 (1998)
4. Takahashi, K. & Yamanaka, S. 定義された因子によるマウス胚性および成人線維芽細胞培養からの多能性幹細胞の誘導。 Cell 126, 663–676 (2006)
5. Nagy, A., Rossant, J., Nagy, R., Abramow-Newerly, W. & Roder, J. C. 早期継代胚性幹細胞からの完全に細胞培養由来のマウス誘導。 Proc. Natl Acad. Sci. USA 90, 8424–8428 (1993)
(英文)
6. Niwa, H. How is pluripotency determined and maintained? Development 134, 635–646 (2007)
7. Beddington, R. S. & Robertson, E. J. An assessment of the developmental potential of embryonic stem cells in the midgestation mouse embryo. Development 105, 733–737 (1989)
8. Quinn, J., Kunath, T. & Rossant, J. Mouse trophoblast stem cells. Methods Mol. Med. 121, 125–148 (2006)
9. Tanaka, S., Kunath, T., Hadjantonakis, A. K., Nagy, A. & Rossant, J. Promotion of trophoblast stem cell proliferation by FGF4. Science 282, 2072–2075 (1998)
10. Tanaka, T. S. et al. Gene expression profiling of embryo-derived stem cells reveals candidate genes associated with pluripotency and lineage specificity. Genome Res. 12, 1921–1928 (2002)
6. Niwa, H. 多能性は如何に決定され、維持されるか? Development 134, 635–646 (2007)
7. Beddington, R. S. & Robertson, E. J.妊娠中期マウス胚における胚性幹細胞の発生能の評価。 Development 105, 733–737 (1989)
8. Quinn, J., Kunath, T. & Rossant, J. マウス栄養膜幹細胞。 Methods Mol. Med. 121, 125–148 (2006)
9. Tanaka, S., Kunath, T., Hadjantonakis, A. K., Nagy, A. & Rossant, J. FGF4による栄養膜幹細胞増殖の促進。 Science 282, 2072–2075 (1998)
10. Tanaka, T. S. et al. 胚由来幹細胞の遺伝子発現プロファイリングは、多分化能および系統特異性に関連する候補遺伝子を明らかにする。Genome Res. 12, 1921–1928 (2002)
(英文)
11. Klaffky, E. et al. Trophoblast-specific expression and function of the integrin alpha 7 subunit in the peri-implantation mouse embryo. Dev. Biol. 239, 161–175 (2001)
12. Russ, A. P. et al. Eomesodermin is required for mouse trophoblast development and mesoderm formation. Nature 404, 95–99 (2000)
13. Niwa, H. et al. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell 123, 917–929 (2005)
14. Mikkelsen, T. S. et al. Dissecting direct reprogramming through integrative genomic analysis. Nature 454, 49–55 (2008)
15. van Oosten, A. L., Costa, Y., Smith, A. & Silva, J. C. JAK/STAT3 signalling is sufficient and dominant over antagonistic cues for the establishment of naive pluripotency. Nature Commun. 3, 817 (2012)
16. Yang, J. et al. Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell Stem Cell 7, 319–328 (2010)
17. Ying, Q. L. et al. The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 (2008)
11. Klaffky, E. et al. 移植前後のマウス胚におけるインテグリンα7サブユニットの栄養膜特異的発現および機能。 Dev. Biol. 239, 161–175 (2001)
12. Russ, A. P. et al. マウス栄養膜の発達および中胚葉形成にEomesoderminが必要。 Nature 404, 95–99 (2000)
13. Niwa, H. et al. Oct3 / 4とCdx2との間の相互作用が栄養外胚葉の分化を決定する。 Cell 123, 917–929 (2005)
14. Mikkelsen, T. S. et al.統合的ゲノム分析による直接再プログラミングの解明。 Nature 454, 49–55 (2008)
15. van Oosten, A. L., Costa, Y., Smith, A. & Silva, J. C.JAK / STAT3シグナル伝達は、原初多能性の確立に関する対立的開始合図を凌いで、十分であり、かつ圧倒的である。 Nature Commun. 3, 817 (2012)
16. Yang, J. et al. Stat3活性化は、基底多能性への再プログラミングを制限している。 Cell Stem Cell 7, 319–328 (2010)
17. Ying, Q. L. et al.胚性幹細胞の基底状態は自己再生する。 Nature 453, 519–523 (2008)
(英文)
18. Bernstein, B. E. et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006)
19. Meek, S. et al. Tuning of β-catenin activity is required to stabilize self-renewal of rat embryonic stem cells. Stem Cells 31, 2104–2115 (2013)
20. Chen, Y., Blair, K. & Smith, A. Robust self-renewal of rat embryonic stem cells requires fine-tuning of glycogen synthase kinase-3 inhibition. Stem Cell Rep. 1, 209–217 (2013)
21. Debeb, B. G. et al. Isolation of Oct4-expressing extraembryonic endoderm precursor cell lines. PLoS ONE 4, e7216 (2009)
22. Macfarlan, T. S. et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63 (2012)
23. Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010)
18. Bernstein, B. E. et al. 二価クロマチン構造は、胚性幹細胞における重要な発生遺伝子をマークする。 Cell 125, 315–326 (2006)
19. Meek, S. et al. ラット胚性幹細胞の自己再生を安定化させるためにβ-カテニン活性の調整が必要である。 Stem Cells 31, 2104–2115 (2013)
20. Chen, Y., Blair, K. & Smith, A. ラット胚性幹細胞の強力な自己再生には、グリコーゲンシンターゼキナーゼ-3阻害の微調整が必要である。 Stem Cell Rep. 1, 209–217 (2013)
21. Debeb, B. G. et al. Oct4発現胚体外胚葉前駆細胞株の単離。 PLoS ONE 4, e7216 (2009)
22. Macfarlan, T. S. et al. 胚性幹細胞の効力は、内因性レトロウイルス活性と共に変動する。 Nature 487, 57–63 (2012)
23. Anders, S. & Huber, W. 配列カウントデータのための特異的発現分析。 Genome Biol. 11, R106 (2010)
(英文)
Acknowledgements
We thank S. Nishikawa and N. Love for discussion and M. Ohgushi, S. Kuraku, M. Eiraku, S. Ohtsuka and K. Kakiguchi for help with experimental planning, material preparation and analyses. Financial support for this research was provided by Intramural RIKEN Research Budget (H.O., T.W. and Y.S.), a Scientific Research in Priority Areas (20062015) to T.W., the Network Project for Realization of Regenerative Medicine to Y.S., and Department of Anesthesiology, Perioperative and Pain Medicine at Brigham and Women’s Hospital to C.A.V.
謝辞
私たちは、討論に関してS. Nishikawa と N. Loveに、実験計画、材料の準備と分析の援助に関して、M. Ohgushi、S. Kuraku、M. Eiraku、S. Ohtsuka 及び K. Kakiguchiに謝意を表する。本研究の資金援助は、理研内の理研研究予算(小保方晴子、若山照彦、笹井芳樹)、若山輝彦への優先分野の科学研究(20062015)、笹井芳樹への再生医療の実現のためのネットワークプロジェクト、及びチャールズ・A・ヴァカンティへのブリガム・ウィミンズ病院麻酔部門術後処置と痛み治療科によって提供された。
(英文)
Author information
Affiliations
1. Laboratory for Cellular Reprogramming, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan
Haruko Obokata &
Yukari Terashita
2. Laboratory for Genomic Reprogramming, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan
Haruko Obokata,
Mikiko Tokoro,
Yukari Terashita &
Teruhiko Wakayama
著者情報
所属
1.細胞リプログラミングのための研究室、理化学研究所発生生物学センター、神戸650-0047、日本
Haruko Obokata &
Yukari Terashita
2.ゲノムリプログラミングのための研究室、理化学研究所発生生物学センター、神戸650-0047、日本
Haruko Obokata,
Mikiko Tokoro,
Yukari Terashita &
Teruhiko Wakayama
(英文)
3. Laboratory for Tissue Engineering and Regenerative Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts 02115, USA
Haruko Obokata &
Charles A. Vacanti
4. Laboratory for Organogenesis and Neurogenesis, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan
Yoshiki Sasai &
Nozomu Takata
5. Laboratory for Pluripotent Stem Cell Studies, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan
Hitoshi Niwa
3.組織工学および再生医療のための研究室、ブリガム・ウィミンズ病院、ハーバード大学医学部、ボストン、マサチューセッツ州02115、米国
Haruko Obokata &
Charles A. Vacanti
4.器官形成および神経発生のための研究室、理化学研究所発生生物学センター、神戸650-0047、日本
Yoshiki Sasai &
Nozomu Takata
5.多能性幹細胞学のための研究室、理化学研究所発生生物学センター、神戸650-0047、日本
Hitoshi Niwa
(英文)
6. Genome Resource and アnalysis Unit, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan
Mitsutaka Kadota &
Munazah Andrabi
7. Electron Microscopy Laboratory, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan
Shigenobu Yonemura
8. Faculty of Life and Environmental Sciences, University of Yamanashi, Yamanashi 400-8510, Japan
Teruhiko Wakayama
6.ゲノムリソース解析ユニット、理化学研究所発生生物学センター、神戸650-0047、日本
Mitsutaka Kadota &
Munazah Andrabi
7.電子顕微鏡検査室、理化学研究所発生生物学センター、神戸650-0047、日本
Shigenobu Yonemura
8.生命環境科学学部、山梨大学、山梨400-8510、日本
Teruhiko Wakayama
(英文)
Contributions
H.O. and Y.S. wrote the manuscript. H.O., Y.S., M.K., M.A., N.T., S.Y. and T.W. performed experiments, and M.T. and Y.T. assisted with H.O.’s experiments. H.O., Y.S., H.N., C.A.V. and T.W. designed the project.
Competing financial interests
The authors declare no competing financial interests.
Corresponding authors
Correspondence to:
Haruko Obokata or
Teruhiko Wakayama or
Yoshiki Sasai
貢献
H.O. 、Y.S. が原稿執筆し、 H.O.、Y.S.、M.K.、M.A.、N.T.、S.Y. 、T.W. が実験実施し、M.T. 、Y.T. がH.O.の実験補助し、 H.O.、Y.S.、H.N.、C.A.V. T.W. がプロジェクトを設計した。
利益相反
著者らは、利益相反の無いことを宣言する。
責任著者
下記へ連絡
Haruko Obokata or
Teruhiko Wakayama or
Yoshiki Sasai
(英文)
RNA-seq and ChIP-seq files have been submitted to the NCBI BioSample databases under accessions SAMN02393426, SAMN02393427, SAMN02393428, SAMN02393429, SAMN02393430, SAMN02393431, SAMN02393432, SAMN02393433, SAMN02393434 and SAMN02393435.
RNA-seqおよびChIP-seqファイルは、SAMBI02393426、SAMN02393427、SAMN02393428、SAMN02393429、SAMN02393430、SAMN02393431、SAMN02393431、SAMN02393432、SAMN02393433、SAMN02393434およびSAMN02393435としてNCBI BioSampleデータベースに提出されている。
- 2019/05/14(火) 09:22:27|
- レター論文
-
-
| コメント:0
(英文)
Methods
Animal studies
Research involving animals complied with protocols approved by the Harvard Medical School/Brigham and Women’s Hospital Committee on Animal Care, and the Institutional Committee of Laboratory Animal Experimentation of the RIKEN Center for Developmental Biology.
方法
動物研究
動物を対象とした研究は、ハーバード大学医学部/ブリガム・ウィミンズ病院の動物実験委員会、理化学研究所発生生物学センターの研究所実験動物組織委員会の承認を受けた。
(英文)
Cell culture
STAP cells were generated from low-pH-treated CD45+ cells, followed by culture in B27 + LIF medium for 7 days, as described1. For Fgf4-induced stem-cell line establishment, STAP cell clusters were transferred to Fgf4-containing trophoblast stem-cell medium9 on MEF feeder cells in 96-well plates. In most cases (40 out of 50 experiments), colonies grew in 10–50% of wells in 96-well plates. In minor cases (10 out of 50 experiments), no colony growth was observed and/or only fibroblast-like cells appeared. The cells were subjected to the first passage during days 7–10 using a conventional trypsin method. Subsequent passages were performed at a split ratio of 1:4 every third day before they reached subconfluency.
STAP stem-cell lines were established as described1. STAP spheres were transferred to ACTH-containing medium1 on MEF feeder cells (several spheres, up to a dozen spheres, per well of 96-well plates). Four to seven days later, the cells were subjected to the first passage using a conventional trypsin method, and suspended cells were plated in ES maintain medium containing 20% FBS. Subsequent passaging was performed at a split ratio of 1:10 every second day before they reached subconfluency.
細胞培養
STAP細胞は、低pH処理CD45 陽性細胞から生成され、続いて既述の通り[1]、B27 + LIF培地中で7日間培養された。 Fgf4誘導幹細胞株樹立ために、STAP細胞クラスターを、96ウェルプレート中のMEFフィーダー細胞上のFgf4含有栄養膜幹細胞培地[9]に移した。 ほとんどの場合(50回の実験のうち40回)、96ウェルプレートの10~50%のウェルでコロニーが増殖した。 稀に(50回の実験のうち10回)、コロニーの成長は観察されないか、または、線維芽細胞様細胞のみが出現した。 細胞は、従来のトリプシン法を用いて7~10日目の間に第1継代に供された。 その後の継代は、サブコンフルに達する前に、3日ごとに1:4の分裂比で実行された。
STAP幹細胞株は既述の通りに[1]樹立された。 STAPスフィアを、MEFフィーダー細胞上のACTH含有培地[1]に移した(96ウェルプレートのウェル当たり、数スフィア、最大12スフィア)。 4~7日後、通常のトリプシン法を用いて細胞を第1継代に供し、懸濁した細胞を20%FBSを含むES維持培地に播種した。 その後の継代は、サブコンフルに達する前に2日ごとに1:10の分割比で行われた。
(英文)
Chimaera mouse generation and analyses
For production of diploid and tetraploid chimaeras with STAP cells, STAP stem cells and Fgf4-induced stem cells, diploid embryos were obtained from ICR strain females. Tetraploid embryos were produced by electrofusion of 2-cell embryos. Because trypsin treatment of donor samples turned out to cause low chimaerism, STAP spherical colonies were cut into small pieces using a microknife under microscopy, and small clusters of STAP cells were then injected into day-4.5 blastocysts by a large pipette. Next day, the chimaeric blastocysts were transferred into day-2.5 pseudopregnant females.
キメラマウスの作成と分析
STAP細胞、STAP幹細胞およびFgf4誘導幹細胞からの、二倍体および四倍体キメラの作成のために、二倍体胚をICR系統の雌から得た。 四倍体胚は2細胞胚の電気融合によって作成された。 ドナー試料のトリプシン処理は低キメニズムを引き起こすことが判明したので、顕微鏡下でマイクロナイフを用いてSTAP球状コロニーを小片に切断し、STAP細胞の小さなクラスターを大型ピペットで4.5日の胚盤胞に注入した。 翌日、キメラ胚盤胞を2.5日の偽妊娠雌に移した。
(英文)
In vivo differentiation assay
1 × 10(to the power of 5) cells of Fgf4-induced stem-cell-derived ES-like cells were injected subcutaneously into the dorsal flanks of 4-week-old NOD/SCID mice. Six weeks later, the implants were collected and histologically analysed. The implants were fixed with 10% formaldehyde, embedded in paraffin, and routinely processed into 4-μm-thick sections. Sections were stained with haematoxylin and eosin. So far, we have not investigated whether Fgf4-induced stem cells form tumours such as teratomas and yolk sac tumours in vivo.
体内分化実験
Fgf4誘導幹細胞由来ES様細胞の1×10の5乗個の細胞を4週齢NOD / SCIDマウスの背側腹部に皮下注射した。 6週間後インプラントを収集し、組織学的に分析した。 インプラントを10%ホルムアルデヒドで固定し、パラフィンに包埋し、普通のやり方で4μmの厚さに加工した。 切片をヘマトキシリンおよびエオシンで染色した。 これまでのところ、Fgf4誘導幹細胞が体内で奇形腫や卵黄嚢腫瘍などの腫瘍を形成するかどうかについては検討していない。
(英文)
Immunostaining
Cells were fixed with 4% PFA for 15 min and, after permeabilization, with 0.5% Triton X-100 and then incubated with primary antibodies: anti-H3K27me3 (Millipore; 1:300), anti-Oct3/4 (Santa Cruz Biotechnology; 1:300), anti-Nanog (eBioscience; 1:300), anti-KLF4 (R&D System; 1:300), anti-Esrrβ (R&D System; 1:300) and integrin α7 antibody (R&D system; 1:200). After overnight incubation, bound antibodies were visualized with a secondary antibody conjugated to Alexa546 (Molecular Probes). Nuclei were stained with DAPI (Molecular Probes).
免疫染色
細胞を4%PFAで15分間固定し、0.5%Triton X-100で透過処理後、以下の一時抗体で保温維持した:抗H3K27me3(Millipore; 1:300)、抗Oct3 / 4(Santa Cruz Biotechnology;1:300)、抗Nanog(eBioscience; 1:300)、抗KLF4(R&D System; 1:300)、抗Esrrβ(R&D System; 1:300)およびインテグリンα7抗体(R&D system; 1:200 )。 一晩保温維持した後、結合した抗体をAlexa546(Molecular Probes)を配合した二次抗体で視覚化した。 核をDAPI(Molecular Probes)で染色した。
(英文)
RNA preparation and RT–PCR analysis
RNA was isolated with the RNeasy Mini kit (Qiagen). Reverse transcription was performed with the SuperScript III First Strand Synthesis kit (Invitrogen). Power SYBR Green Mix (Roche Diagnostics) was used for PCR amplification, and samples were run on a Lightcycler-II Instrument (Roche Diagnostics). The primer sets for each gene are listed in Supplementary Table 1.
RNA調製およびRT-PCR分析
RNeasy Miniキット(Qiagen)を用いてRNAを単離した。 逆転写は、SuperScript IIIファーストストランド合成キット(Invitrogen)を用いて行った。 Power SYBR Green Mix(Roche Diagnostics)をPCR増幅に使用し、サンプルをLightcycler-II Instrument(Roche Diagnostics)で実行した。 各遺伝子のプライマーセットを補遺表1に示す。
(英文)
Inhibitor assay
For JAK inhibitor assay, Fgf4-induced stem cells were cultured without feeders for 48 h in trophoblast stem-cell culture medium supplemented with 0.6 μM JAK inhibitor (CalBiochem, 420097). As a control, ES cells were also cultured for 48 h in ES medium supplemented with 0.6 μM JAK inhibitor. After the JAK inhibitor treatment, cells were collected and their gene expression was analysed by RT–PCR. For MEK inhibitor assay, dissociated Fgf4-induced stem cells were plated in either LIF containing ES medium supplemented with 1 μM MEK inhibitor (PD025901) or FGF4 containing trophoblast stem cell medium supplemented with 1 μM MEK inhibitor for 48 h. As controls, dissociated Fgf4-induced stem cells were co-plated with 5% or 50% of ES cells into the same culture conditions. After the MEK inhibitor treatment, colonies that formed in each culture condition were counted.
阻害剤実験
JAK阻害剤実験のために、0.6μMのJAK阻害剤(CalBiochem、420097)を補充した栄養膜幹細胞培養培地中でFgf4誘導幹細胞をフィーダーなしで48時間培養した。 コントロールとして、ES細胞を0.6μMのJAK阻害剤を補充したES培地で48時間培養した。 JAK阻害剤処理の後、細胞を収集し、それらの遺伝子発現をRT-PCRによって分析した。 MEK阻害剤アッセイのために、解離したFgf4誘発幹細胞を1μMのMEK阻害剤(PD025901)を補充したLIF含有ES培地、または1μMのMEK阻害剤を補充した栄養膜幹細胞培地を含むFGF4培地に48時間播種した。コントロールとして、解離したFgf4誘導性幹細胞を、5%または50%のES細胞と共培養した。 MEK阻害剤処理後、各培養条件で形成されたコロニーを数えた。
(英文)
FACS sorting
Fgf4-induced stem cells were dissociated into single cells and were suspended in 0.5% BSA PBS. Suspended cells were Fc-blocked by treatment with 1 μg of mouse IgG per 10(to yhe power of 5) cells for 15 min at room temperature. PE-conjugated integrin α7 antibody (R&D system, FAB3518P, dilution at 1:10) was added into cell suspension, and cells were incubated for 30 min on ice. Finally, cells were rinsed with PBS three times and propidium iodide was added for dead cell elimination. As a control, Fgf4-induced stem cells in a separate tube were treated with PE-labelled rat IgG2B antibody. Integrin α7-positive and -dim cells were sorted by FACS aria II (BD).
FACSソーティング
Fgf4誘導幹細胞を単細胞に解離させ、0.5%BSA PBSに懸濁させた。 懸濁した細胞を、室温で15分間、10の5乗細胞あたり1μgのマウスIgGで処理することによってFc-遮断した。PE結合インテグリンα7抗体(R&Dシステム、FAB3518P、1:10希釈)を細胞懸濁液に加え、細胞を氷上で30分間インキュベートした。 最後に、細胞をPBSで3回すすぎ、死細胞除去のためにヨウ化プロピジウムを添加した。 コントロールとして、別のチューブ中のFgf4誘導幹細胞をPE標識ラットIgG2B抗体で処理した。 インテグリンα7陽性および 弱発現細胞をFACS aria II(BD)で選別した。
(英文)
RNA sequencing and ChIP sequencing analyses
RNA-sequencing of cell lines was performed with biological duplicate samples. Total RNA was extracted from T cells by the RNasy mini kit (Qiagen). RNA-seq libraries were prepared from 1 μg total RNAs following the protocol of the TruSeq RNA Sample Prep kit (Illumina) and subjected to the deep sequencing analysis with Illumina Hi-Seq1000. A cluster tree diagram of various cell types was obtained from hierarchical clustering of global expression profiles (log2 FPKM of all transcripts; FPKM, fragments per kilobase of transcript per million mapped reads). Complete linkage method applied to 1 - r (r = Pearson’s correlation between profiles) was used for generating the tree and 1,000 cycles of bootstrap resampling were carried out to obtain statistical confidence score in % units (also called AU P values). For the analysis that included morula and blastocyst embryos (only small amounts of RNA can be obtained from them), we used pre-amplification with the SMARTer Ultra Low RNA kit for Illumina Sequencing (Clontech Laboratories). Differentially expressed genes were identified by the DESeq package23.
RNAシークエンシングおよびChIPシークエンシング解析
生物学的二重試料を用いて細胞系のRNA配列決定を行った。全RNAをRNasyミニキット(Qiagen)によりT細胞から抽出した。 RNA-seqライブラリーは、TruSeq RNA Sample Prepキット(イルミナ)のプロトコールに従って1μgの全RNAから調製し、Illumina Hi-Seq1000で深い配列決定分析を行った。全発現プロファイルの(全転写物のlog2 FPKM; FPKMは、マップされた百万リードに対して、転写物のキロベースあたりの断片数を意味する)階層的クラスタリングから、様々な細胞型のクラスターツリー図を得た。ツリーを作成するために、1-r(r=プロファイル間のピアソン相関係数)に適用された完全リンケージ法を使用し、%単位(AU P値とも呼ばれる)で統計的信頼スコアを得るために1,000サイクルのブートストラップ再サンプリングを実施した。桑実胚および胚盤胞胚を含む分析(少量のRNAしか得られない)に関しては、Illumina Sequencing(Clontech Laboratories)用のSMARTer Ultra Low RNAキットで事前増幅を行った。異様発現遺伝子は、DESeqパッケージ23によって同定された。
(英文)
ChIP-seq libraries were prepared from 20 ng input DNAs, 1 ng H3K4me3 ChIP DNAs, or 5 ng H3K27me3 ChIP DNAs using the KAPA Library Preparation kit (KAPA Biosystems). TruSeq adaptors were prepared in-house by annealing a TruSeq universal oligonucleotide and each of index oligonucleotides (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′, and 5′-GATCGGAAGAGCACACGTCTGAACTCCAGTCACXXXXXXATCTCGTATGCCGTCTTCTGCTTG-3′; where X represents index sequences).
Chromatin immunoprecipitation was performed as follows. Cells were fixed in PBS(-) containing 1% formaldehyde for 10 min at room temperature. Glycine was added to a final concentration of 0.25 M to stop the fixation. After washing the cells twice in ice-cold PBS(-), cells were further washed in LB1 (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100) and LB2 (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA). Cells were then re-suspended in lysis buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS). Lysates were prepared by sonication using COVARIS S220 in a mini tube at duty cycle = 5%, PIP = 70, cycles per burst = 200, and the treatment time of 20 min. Lysates from 2 × 106 cells were diluted in ChIP dilution buffer (16.7 mM Tris-HCl pH 8.0, 167 mM NaCl, 1.2 mM EDTA, 1.1% Triton X-100, 0.01% SDS). ChIP was performed using sheep anti-mouse IgG beads (Invitrogen) or protein A beads (Invitrogen) coupled with anti-histone H3K4me3 antibody (Wako, catalogue no. 307-34813) or anti-histone H3K27me3 antibody (CST, catalogue no. 9733), respectively.
ChIP-seqライブラリーを、20ngの入力DNA、1ngのH3K4me3 ChIP DNA、または5ngのH3K27me3 ChIP DNAから、KAPAライブラリー調製キット(KAPA Biosystems)を用いて調製した。 TruSeqアダプターは、TruSeqユニバーサルオリゴヌクレオチドおよび各インデックスオリゴヌクレオチド(5'-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGCGCTCTTCCGATCT-3 'および5'-GATCGGAAGAGCACACGTCTGAACTCCAGTCACXXXXXXATCTCGTATGCCGTCTTCTGCTTG-3';ここで、Xはインデックス配列を表す)のアニーリングによって社内で調製した。
クロマチン免疫沈降は以下のように行った。細胞を1%ホルムアルデヒドを含むPBS( - )中で室温で10分間固定した。グリシンを最終濃度0.25Mになるように加えて固定を停止させた。氷冷PBS( - )で細胞を2回洗浄した後、LB1(50mM HEPES-KOH pH7.5,140mM NaCl、1mM EDTA、10%グリセロール、0.5%NP-40,0.25%Triton X-100)およびLB2(10mMトリス-HCl pH8.0,200mM NaCl、1mM EDTA、0.5mM EGTA)で洗浄した。次いで、細胞を溶解緩衝液(50mMトリス-HCl pH8.0,10mM EDTA、1%SDS)に再懸濁した。溶解物は、デューティーサイクル= 5%、PIP = 70、バースト当たりのサイクル= 200、および20分の処理時間でミニチューブ中でCOVARIS S220を使用する超音波処理によって調製した。 2×10 の6乗細胞からの溶解物をChIP希釈緩衝液(16.7mMトリス-HCl pH8.0,167mM NaCl、1.2mM EDTA、1.1%Triton X-100,0.01%SDS)で希釈した。 ChIPは、抗ヒストンH3K4me3抗体(Wako、カタログ番号307-34813)または抗ヒストンH3K27me3抗体(CST、カタログ番号9733)と結合したヒツジ抗マウスIgGビーズ(Invitrogen)またはプロテインAビーズ(Invitrogen) )を使って実施された。
(英文)
After 4–6 h of incubation in a rotator at 4 °C, beads were washed five times in low-salt wash buffer (20 mM Tris HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), and three times in high-salt wash buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS). Target chromatin was eluted off the beads in elution buffer (10 mM Tris-HCl pH 8.0, 300 mM NaCl, 5 mM EDTA, 1% SDS) at room temperature for 20 min. Crosslink was reversed at 65 °C, and then samples were treated with RNaseA and proteinase K. The prepared DNA samples were purified by phenol-chloroform extraction followed by ethanol precipitation and dissolved in TE buffer.
4℃で回転子中で4~6時間保温した後、ビーズを低塩洗浄緩衝液(20mM Tris HCl pH8.0,150mM NaCl、2mM EDTA、1%Triton X-100,0.1 %SDS)で5度洗浄し、高塩洗浄緩衝液(20mM Tris-HCl pH8.0,500mM NaCl、2mM EDTA、1%Triton X-100,0.1%SDS)中で3度洗浄した。 溶出緩衝液(10mMトリス-HCl pH8.0,300mM NaCl、5mM EDTA、1%SDS)中、室温で20分間、標的クロマチンをビーズから溶出させた。 65℃で架橋を逆転させた後、サンプルをRNaseAおよびプロテイナーゼKで処理した。調製したDNAサンプルをフェノール - クロロホルム抽出および続いてエタノール沈殿により精製し、TE緩衝液に溶解した。
- 2019/05/14(火) 09:13:57|
- レター論文
-
-
| コメント:0
(英文)
Figure 2: Fgf4 treatment induces some trophoblast-lineage character in STAP cells.
a, Schematic of Fgf4 treatment to induce Fgf4-induced stem cells from STAP cells. b, Fgf4 treatment promoted the generation of flat cell clusters that expressed Oct4-GFP at moderate levels (right). Top and middle: days 1 and 7 of culture with Fgf4, respectively. Bottom: culture after the first passage. Scale bar, 50 μm. c, d, Immunostaining of Fgf4-induced cells with the trophoblast stem cell markers integrin α7 (c) and eomesodermin (d). Scale bar, 50 μm. e, qPCR analysis of marker expression. f, g, Placental contribution of Fgf4-induced stem cells (FI-SCs) (genetically labelled with constitutive GFP expression). Scale bars: 5.0 mm (f (left panel) and g); 50 µm (f, right panel). In addition to placental contribution, Fgf4-induced stem cells contributed to the embryonic portion at a moderate level (g).
h, Quantification of placental contribution by FACS analysis. Unlike Fgf4-induced cells, ES cells did not contribute to placental tissues at a detectable level. i, Cluster tree diagram from hierarchical clustering of global expression profiles. Red, approximately unbiased P values. j, qPCR analysis of Fgf4-induced cells (cultured under feeder-free conditions) with or without JAK inhibitor (JAKi) treatment for pluripotent marker genes. k, qPCR analysis of FI-SCs with or without JAK inhibitor (JAKi) treatment for trophoblast marker genes. Values are shown as ratio to the expression level in ES cells (j) or trophoblast stem cells (k). ***P < 0.001; NS, not significant; t-test for each gene between groups with and without JAK inhibitor treatment. n = 3. Statistical significance was all the same with three pluripotency markers. None of the trophoblast marker genes showed statistical significance. Error bars represent s.d.
図2:Fgf4処理は、STAP細胞においていくつかの栄養膜関連性質を誘導する。
a,STAP細胞からのFgf4誘導幹細胞を誘導するFgf4処置の概略図。 b,Fgf4処理は中程度のレベルでOct4-GFPを発現する扁平細胞塊の生成を促進した(右)。上段と中段:Fgf4によるそれぞれ1日目と7日目の培養。下段:最初の継代後の培養。スケールバーは50μm。 c, d, 栄養膜幹細胞マーカーであるインテグリンα7(c)とエオメソダミン(d)のFgf4誘導細胞の免疫染色。スケールバー、50μm。 e,マーカー発現の定量PCR分析。 f, g,Fgf4誘導幹細胞(FI-SCs)の胎盤の寄与(構造的GFP発現で遺伝子操作標識されている)。スケールバー:5.0mm(fの左パネルおよびg);50µm(fの右パネル)。胎盤の寄与に加えて、Fgf4誘導幹細胞は、中程度のレベルでの胚部分にも貢献した(g)。
h, FACS分析による胎盤貢献の定量化。 Fgf4誘導細胞とは異なり、ES細胞は検出可能なレベルで胎盤組織に寄与しなかった。 i, 網羅的発現特性階層的分類による分類系統樹図。赤字はほぼ公平なP値。 j,多能性マーカー遺伝子のためのJAK阻害剤(JAKi)処置の有無で分けられた(フィーダー無し条件下で培養された)Fgf4誘導細胞のqPCR分析。 k,栄養膜幹細胞マーカー遺伝子へのJAK阻害剤(JAKi)処置の有無で分けられたFI-SCsのqPCR分析。値はES細胞(j)または栄養膜幹細胞(k)の中の発現レベルに対する比として示されている。 *** P <0.001; NS、重要でない;JAK阻害剤処理の有無によるグループ間の各遺伝子についてのt検定。 N = 3。三つの多能性マーカーの統計的有意性はまったく同じであった。栄養膜幹細胞のマーカー遺伝子のいずれも統計的有意性を示さなかった。エラーバーは±標準偏差を表す。
(英文)
These Fgf4-induced cells with trophoblast marker expression could be expanded efficiently in the presence of Fgf4 by passaging for more than 30 passages with trypsin digestion every third day. Hereafter, these proliferative cells induced from STAP cells by Fgf4 treatment are referred to as Fgf4-induced stem cells. This type of derivation into trophoblast-stem-like cells is not common with ES cells (unless genetically manipulated)<13> or STAP stem cells.
In the blastocyst injection assay, unlike STAP stem cells, the placental contribution of Fgf4-induced stem cells (cag-GFP-labelled) was observed with 53% of embryos (Fig. 2f, g; n = 60). In the chimaeric placentae, Fgf4-induced stem cells typically contributed to ~10% of total placental cells (Fig. 2h and Extended Data Fig. 2a, b).
栄養膜マーカー発現を伴うこれらのFgf4誘導細胞は、Fgf4存在下で、三日毎のトリプシン分離処理で30継代以上の間継代することにより効率的に増殖することができた。それより後、Fgf4処理によりSTAP細胞から誘導されたこれらの増殖細胞はFgf4誘導幹細胞と呼ぶことにする。栄養膜幹様細胞様細胞へのこのタイプの派生物は、ES細胞とも(遺伝子操作しない限り)、STAP幹細胞とも同じものではない。
胚盤胞注入実験において、STAP幹細胞とは異なり、Fgf4誘導幹細胞(CAG-GFP標識)の胎盤寄与は胚の53%で観察された(図2f,g; n=60)。キメラ胎盤では、Fgf4誘導幹細胞は典型的な例では全胎盤細胞の最大10%に貢献する(図2h及び拡張データ図2a,b)。
(英文)
Despite their similarities, we noted that Fgf4-induced stem cells also possessed some critical differences compared with blastocyst-derived trophoblast stem cells. First, Fgf4-induced stem cells exhibited moderate GFP signals and expressed a moderate level of Oct4 (Fig. 2b; moderate and low levels of immunostaining signals were also seen for Oct4 and Nanog proteins, respectively; Extended Data Fig. 2c), unlike conventional trophoblast stem cells that have little Oct4 expression (Fig. 2e). Second, unlike trophoblast stem cells, blastocyst-injected Fgf4-induced stem cells also contributed to embryonic tissues (in all cases that involved chimaeric placentae; n = 32), although the extent of contribution was generally modest (Fig. 2g).
Third, immunostaining revealed that the level of Cdx2 protein accumulation in the nuclei of Fgf4-induced stem cells was marginal as compared to the cytoplasmic level, although the transcript expression level was substantial (Fig. 2e). This may suggest complex and dynamic post-transcriptional regulations for this key transcription factor in Fgf4-induced stem cells (a similar situation was seen for STAP cells, in which clear nuclear localization was not observed for either Cdx2 or Eomes, despite substantial expression of their transcripts). Fourth, in the absence of Fgf4, Fgf4-induced stem cells gradually died in 7–10 days and did not differentiate into large and multi-nuclear cells, unlike trophoblast stem cells (Extended Data Fig. 2d).
それらの類似性にもかかわらず、我々はFgf4誘導幹細胞は、胚盤胞由来栄養膜幹細胞と比較していくつかの重要な違いを持っていたことを指摘した。第一に、従来の栄養膜幹細胞がOct4蛋白発現が皆無であるのに対して(図2e)、Fgf4誘導幹細胞は適度なGFPシグナルを示し、そして適度なレベルOct4蛋白を発現した(図2b;中程程度および低いレベルで免疫染色シグナルが、それぞれ、Oct4とNanog蛋白質について見られた;拡張データ図2c)。第二に、栄養膜幹細胞とは異なり、胚盤胞に注入したFgf4誘導幹細胞は、寄与の程度は、一般的に控えめではあるものの(図2g)、胚組織にも寄与した(キメラ胎盤を含むすべてのケースにおいて;n = 32)、
第三に、免疫染色は、細胞質内のレベルと比較して、転写産物発現レベルが実体的であったのに対して(図2e)、Fgf4誘導幹細胞の核内のCdx2蛋白質の蓄積レベルがわずかであることを明らかにした。これはFgf4誘導幹細胞の主要な転写因子にとっての複雑で動的な転写後規制を示唆しているのかもしれない(同様の状況がSTAP細胞に見られた。STAP細胞の中では、それらの転写の実態的表出があるにも関わらず、Cdx2またはEomesのいずれについても明確な核局在が観察されなかった。第四に、栄養膜幹細胞(拡張データ図2d)とは異なり、Fgf4非存在下では、Fgf4誘導幹細胞は徐々に7日から10日内に死滅し、大きく多能な核細胞に分化しなかった。
(英文)
To investigate the relationship among STAP cells, STAP stem cells, Fgf4-induced stem cells, ES cells and trophoblast stem cells, we performed genome-wide RNA-sequencing analysis (Fig. 2i for dendrogram; Extended Data Figs 3 and 4 for expression analyses of representative genes ; Supplementary Tables 2 and 3 for analysis conditions). Whereas STAP cells formed a cluster with STAP stem cells, Fgf4-induced stem cells, ES cells and trophoblast stem cells and not with the parental CD45+ cells, STAP cells were an outlier to the rest of the cell types in the cluster. In contrast, STAP stem cells were closely clustered with ES cells. Fgf4-induced stem cells formed a cluster with a sub-cluster of ES cells and STAP stem cells, whereas trophoblast stem cells comprised an outlier to this cluster, indicating a close relationship of Fgf4-induced stem cells with these pluripotent cells.
However, as Fgf4-induced stem cells lay between STAP stem cells and trophoblast stem cells in the dendrogram, the possibility of contamination of STAP stem cells in the Fgf4-induced stem-cell population cannot be ruled out. Previous studies have indicated that inner cell mass (ICM)-type pluripotent cells can be removed from culture by treating the culture with a JAK inhibitor <16>(Extended Data Fig. 5a, b). In contrast, the JAK inhibitor treatment had no substantial effect on Oct4-GFP expression in Fgf4-induced stem-cell culture (Extended Data Fig. 5c, d; see Extended Data Fig. 5e, f for control). Expression of neither pluripotency markers (Fig. 2j) nor trophoblast markers (Fig. 2k) was substantially affected, indicating that pluripotency marker expression is unlikely to reflect contaminating STAP stem cells (ICM-type). Consistent with this idea, Fgf4-induced stem cells that were strongly positive for the trophoblast marker Itga7 (a surface marker for trophoblasts but not ES cells) also expressed high levels of Oct4-GFP (Extended Data Fig. 5g).
STAP細胞、STAP幹細胞、Fgf4誘導幹細胞、ES細胞及び栄養膜幹細胞との関係を調べるために、我々は全ゲノムRNA配列決定分析を行った(図2i 系統樹用;拡張データ図3と4 代表的遺伝子の発現解析用;補足表2と3 分析条件用)。STAP細胞は、親のCD45陽性と違って、STAP幹細胞、Fgf4誘導幹細胞、ES細胞および栄養膜幹細胞と同じく細胞塊を形成する一方、STAP細胞は、細胞塊の中で他の細胞タイプとは違って特異であった。対照的に、STAP幹細胞はES細胞に近い細胞塊形成をした。Fgf4誘導幹細胞は、ES細胞およびSTAP幹細胞のサブクラスターに近いクラスターを形成し、一方、栄養膜幹細胞はこのクラスターに異常値を含み、Fgf4誘導幹細胞とこれらの多能性細胞との密接な関係を示した。
しかしながら、Fgf4誘導幹細胞は、樹状図においてSTAP幹細胞と栄養膜幹細胞との間に位置しているので、Fgf4誘導幹細胞集団におけるSTAP幹細胞の混入の可能性を排除できない。先行する研究では、内部細胞塊(ICM)タイプの多能性細胞は、JAK阻害剤で培養を処理することにより培養物から除去することができることを示した(拡張データ図5a,b)。対して、JAK阻害剤処置は、Fgf4誘導幹細胞培養のOct4-GFPの発現には実質的な影響を及ぼさなかった。(拡張データ図5c,d;コントロールとして拡張データ図5e,f参照)。どの多能性マーカー(図2j)や栄養膜マーカー(図2k)も実質的影響を被らなかった。これは多能性マーカー発現がSTAP幹細胞(ICMタイプ)を反映している可能性の低いことを示している。それを証明するように、栄養膜マーカーであるItga7(ES細胞ではなく、栄養膜の表面マーカ)に対して強く陽性であったFgf4誘導幹細胞はOct4-GFP(拡張データ図5g)を高レベルで発現した(拡張データ図5g)。
(英文)
Notably, when cultured in LIF+FBS-containing medium for 4 days, Fgf4-induced stem cells underwent substantial changes in morphology and started to form ES-cell-like compact colonies with strong GFP signals (Fig. 3a). These cells showed expression of pluripotency makers, but not trophoblast markers (Fig. 3b and Extended Data Fig. 6a), and formed teratomas in mice (Extended Data Fig. 6b). These ES-like cells were generated from Fgf4-induced stem cells sorted for strong expression of the trophoblast marker Itga7, but rarely from Itga7-dim cells (Fig. 3c, d).
特に4日間LIF+ FBS含有培地で培養すると、Fgf4誘導幹細胞は、形態学的に実質的な変化を受け、強いGFPシグナル(図3a)を有するES細胞様のコンパクトなコロニーを形成し始めた。これらの細胞は、多能性メーカーの発現を示したが、栄養膜マーカーは示さず(図3b及び拡張データ図6a)、かつマウスにおいてテラトーマを形成した(データ図。拡張図6b)。これらのES様細胞は栄養膜マーカーItga7の強発現しているFgf4誘導幹細胞から生成されたが、まれにItga7-DIM細胞からも生成された(図3c、d)。
(英文)
Figure 3: Fgf4 treatment induces some trophoblast-lineage character in STAP cells.
a, Culture of Oct4-GFP Fgf4-induced cells in LIF + 20% FBS medium. b, qPCR analysis of ES-like cells derived from Fgf4-induced cells for pluripotent marker genes (left) and trophoblast marker genes (right). Values are shown as ratio to the expression level in ES cells (left) or trophoblast stem (TS) cells (right). c, d, Culture of Oct4-GFP Fgf4-induced cells sorted by FACS for strong integrin α7 (Itga7) expression in LIF + 20% FBS medium. d, Formation frequency (shown by percentage) of Oct4-GFP+ colonies from cells plated on gelatin-coated dishes at a clonal density. **P < 0.01; t-test; n = 3. e, f, Culture of Oct4-GFP Fgf4-induced cells (dissociated) in LIF + 20% FBS medium with MEK inhibitor. **P < 0.01; NS, not significant; Tukey’s test; n = 3.
e, No substantial formation of Oct4-GFP+ colonies was seen from Fgf4-induced cells in the presence of MEK inhibitor (left), whereas colonies frequently formed when cells were co-plated with Oct4-GFP ES cells (right; plated cells were 1/20 of Fgf4-induced cells). f, Quantification of colony formation per plated cells (1 × 103 Fgf4-induced cells and/or 1 × 103 ES cells). Unlike Fgf4-induced cells, ES cells formed colonies (regardless of co-plating with FI-SCs) in the presence of MEK inhibitor. Bars and error bars represent mean values and s.d., respectively (b, d, f). Scale bars: 100 μm (a, c, e).
図3:Fgf4処理は、STAP細胞においていくつかの栄養膜の系統特性を誘導する。
a, LIF + 20%FBS培地中でのOct4-GFP FGgf4誘導細胞の培養。 b,多能性マーカー遺伝子(左)と栄養膜マーカー遺伝子(右)のFgf4誘導細胞由来ES様細胞の定量PCR分析。値はES細胞(左)と栄養膜幹細胞(TS)(右)の発現レベルの比で示されている。 c, d, LIF + 20%FBS培地中でインテグリンα7(Itga7)の強発現しているものをFACSソートしたOct4-GFP Fgf4誘導細胞の培養。 d,クローン密度でゼラチンコートディッシュに播種した細胞からのOct4-GFP 陽性コロニーの形成頻度(パーセンテージで示す)。 ** P <0.01;t検定; n = 3 。 e, f, MEK阻害剤入りLIF+ 20%FBS培地での(解離された)Oct4-GFP Fgf4誘導細胞の培養。 ** P <0.01; NS,重要でない;ターキーテスト;n=3。
e,MEK阻害剤(左)の存在下で、Fgf4誘導細胞からOct4-GFP陽性コロニーの実質的な形成は見られなかった。他方、同時播種したOct4-GFP ES細胞(右;播種した細胞はFgf4誘導細胞の1/20だった)からはコロニーが頻繁に形成された。 f,播種した細胞当たりのコロニー形成の定量(1×10の3乗 Fgf4誘導細胞及び/又は1×10の3 乗ES細胞)。 Fgf4誘導細胞とは異なり、ES細胞はMEK阻害剤の存在下で(FI-SC共培養にも関わらず)コロニーを形成した。バーおよびエラーバーはそれぞれ(b,d,f)平均値とsdを表す。スケールバー:100ミクロン(a,c,e)。
(英文)
To confirm further that Fgf4-induced stem cells with a trophoblast-like nature were converted into ES-like cells, rather than just selecting ES-like cells pre-existing in the Fgf4-induced stem cell culture, we examined the effect of the MEK inhibitor PD0325901 on the ES-like cell generation from Fgf4-induced stem cells. Like trophoblast stem cells, Fgf4-induced stem-cell survival is dependent on FGF–MEK signals, and the inhibition of MEK activity caused massive cell death (Extended Data Fig. 6c). However, PD0325901 is also known to be a main effector in 2i medium[17] and to promote ES cell maintenance. Addition of PD0325901 to LIF+FBS-containing medium strongly inhibited the formation of ES-like colonies from Fgf4-induced stem cells (Fig. 3e, left, and Fig. 3f). This inhibition was unlikely to be due to secondary toxic effects from massive cell death of Fgf4-induced stem cells, as colonies formed in the presence of PD0325901 when ES cells were co-plated in the same culture with Fgf4-induced stem cells (Fig. 3e, right, and Fig. 3f).
Collectively, these findings demonstrate that STAP-derived Fgf4-induced stem cells not only express both pluripotency markers and trophoblast genes but also have the potential to convert into ES-like cells when cultured in LIF+FBS-containing medium (Fig. 4a).
単にFgf4誘導幹細胞の培養皿の中にあらかじめ存在していたES様細胞を選別してきたのではなく、栄養膜様の性質を有するFgf4誘導幹細胞がES様細胞に変換したのだということをさらに確認するために、我々は、Fgf4誘導幹細胞からES様細胞への生成に関して、MEK阻害剤、あるいはPD0325901の効果を調べた。栄養膜幹細胞のように、Fgf4誘導幹細胞の生存はFGF-MEKシグナルに依存しており、MEK活性を阻害すると大量の細胞死を引き起こした(拡張データ図6c)。しかし、またPD0325901は2i培地[17]の主要要素で、ES細胞の維持を促進することが知られている。 LIF + FBS含有培地にPD0325901を添加するとFgf4誘導幹細胞からのES様コロニーの形成を強く阻害した(図3e、左、および図3f)。 ES細胞がFgf4誘導幹細胞と同一の培養皿で共培養されていて、PD0325901の存在下でコロニーが形成されたのであるから、この阻害はFgf4誘導幹細胞の大量細胞死からの二次毒性効果の所為ではないようだ( 図3e、右、および図3f)。
まとめると、これらの知見は、STAP由来Fgf誘導幹細胞は、多能性マーカーおよび栄養膜遺伝子の両方を発現するだけでなく、LIF+ FBS含有培地中で培養するとES様細胞に変換する能力をも有することを示している(図4a)。
(英文)
Figure 4: Differentiation potential and epigenetic state of STAP and STAP-derived stem cells.
a, Schematic diagram of stem-cell conversion cultures from STAP cells under different conditions. b, ChIP-sequencing results of histone H3K4 (green) and H3K27 (red) trimethylation at the loci of pluripotent marker genes (left), bivalent pattern genes (middle) and trophoblast marker genes (right). Scale bars indicate 10 kb for pluripotency marker genes and trophoblast marker genes, and 20 kb for bivalent pattern genes.
図4:STAPおよびSTAP由来幹細胞の分化能および後成的(エピジェネティック)状態。
a, 異なる諸条件下でのSTAP細胞から幹細胞への変換培養模式図。 b, 多能性マーカー遺伝子(左)、二価パターン遺伝子(中央)および栄養膜マーカー遺伝子(右)の遺伝子座での、ヒストンH3K4(緑)およびH3K27(赤)のトリメチル化のChIPシーケンシング結果。スケールバーは、多能性マーカー遺伝子と栄養膜マーカー遺伝子は10 kb、二価パターン遺伝子のは20kbを示す。
(英文)
Here we demonstrate that STAP cells, which have a limited self-renewal ability, can be induced to generate two distinct types of robustly self-renewing stem cells—STAP stem cells and Fgf4-induced stem cells—under different culture conditions. Chromatin immunoprecipitation (ChIP) sequencing analysis showed distinct accumulation patterns of modified histone H3 in the two types of STAP-cell-derived stem cells (Fig. 4b). STAP stem cells (as well as STAP cells) had accumulation patterns of H3K4 and H3K27 trimethylation that resembled those of ES cells at the loci of pluripotency marker genes (Oct4, Nanog, Sox2), bivalent pattern genes18 (Gata2, brachyury, Nkx6-2) and trophoblast marker genes (Cdx2, Eomes, Itga7). In contrast, the accumulation patterns in Fgf4-induced stem cells at these loci matched more closely those of trophoblast stem cells, except that low levels of accumulation of H3K4 trimethylation in Oct4 and Nanog and of H3K27 trimethylation in the trophoblast marker genes were observed in Fgf4-induced stem cells but not trophoblast stem cells.
ここでは、限られた自己再生能しか持たないSTAP細胞を、異なる培養条件下で、STAP幹細胞とFgf4誘導幹細胞という、二つの異なるタイプの、確実に自己再生する幹細胞に生成するように誘導することができることを示す。クロマチン免疫沈降法(ChIP)配列決定分析は、二種類のタイプのSTAP細胞由来幹細胞において、修飾されたヒストンH3の厳密な蓄積パターンを示した(図4b)。(STAP細胞と同様に) STAP幹細胞は多能性マーカー遺伝子(Oct4、Nanog、Sox2)、二価パターン遺伝子[18](Gata2, brachyury, Nkx6-2)、および栄養膜マーカー遺伝子(Cdx2の、Eomes、Itga7)の遺伝子座において、ES細胞のものに似たH3K4及びH3K27のトリメチル化蓄積パターンを持つ。対して、FGF4誘導幹細胞における同様の遺伝子座の蓄積パターンはより密接にトロホブラスト幹細胞のものと一致した。ただし、Oct4とNanogにおけるH3K4のトリメチル化、及びトロホブラストのマーカー遺伝子におけるH3K27のトリメチル化のレベルの低さがFgf4誘導細胞では観察されたが、栄養膜幹細胞では見られない。
(英文)
Recent studies have also begun to reveal dynamic regulations in multiple cellular states related to pluripotency. These include reports of co-expression of Oct4 and Cdx2 in rat ES cells maintained in the presence of a GSK-3β inhibitor[19][20] and of Oct4 expression in rat extra-embryonic precursors[21].Another recent study has indicated that conventional ES cell culture also contains a very minor population of Oct4- cells with features resembling those of very early-stage embryos, including bidirectional potential[22]. However, these cells are dissimilar to STAP cells as they are Oct4-, unlike STAP cells and Fgf4-induced stem cells. Our preliminary genome-wide RNA-sequencing analysis indicated that both morulae and blastocysts are outliers to the cluster of STAP and ES cells (Extended Data Fig. 6d–f and Supplementary Tables 4 and 5).
最近の研究はまた、多能性細胞状態の動的制御を明らかにし始めている。これらのなかには、ラットES細胞で、GSK-3βinhibitor[19][20]の存在下で、Oct4およびCdx2の共発現の報告、ラット胚外前駆体[21]におけるOct4発現の報告がある。他の最近の研究では、従来のES細胞培養でも、双方向に向かえる能力を示す、極めて早期段階の胚に似た特徴を有するOct4陰性細胞の僅かな集団が含まれていることを示唆している[22]。しかしながら、これらの細胞は、STAP細胞ともFgf4誘導幹細胞とも異なり、Oct4陰性状態でのSTAP細胞とも似ていない。我々の予備的ゲノムワイドRNA配列決定分析は、桑実胚と胚盤胞の両方ともSTAP細胞とES細胞塊に対して異常値であることを示した(拡張データ図6d-fと補足表4および5)。
(英文)
A key conclusion drawn from this study is that the reprogramming in STAP conversion goes beyond the pluripotent state of ES cells and involves the acquisition of a wider developmental potential related to both ICM- and trophoectoderm-like states. Because of the inability to clone STAP cells from single cells, we must await future technical advancement to examine whether their dual-directional differentiation potential at the population level may reflect one totipotent state at the single-cell level or two different states of STAP cells coexisting (or fluctuating between them) in culture. As for STAP-cell-derived Fgf4-induced stem cells, which can also contribute to both embryonic and placental tissues, our in vitro conversion study combined with inhibitor treatments clearly indicate that the bidirectional potential of Fgf4-induced stem cells is unlikely to reflect the co-presence of separate subpopulations of ES-like and trophoblast-stem-like cells in the culture. Collectively, our study indicates that STAP-based conversion can reprogram somatic cells to acquire not only pluripotency but also the ability of trophoblast differentiation.
この研究から導かれた要所の結論は、STAP変換のリプログラミングは、ES細胞の多能性状態を超えて、インナーセルマスと栄養膜細胞様状態の両方に関連する広い発生能の獲得を伴うことである。単一細胞からのSTAP細胞を増殖することができないために、我々は将来の技術的進歩を待たなければならない。集団レベルでの双方向分化能を単一細胞レベルで、一つの全能性状態なのか、或いはSTAP細胞の二つの異なる状態が培養皿の中に共存しているのかどうか(またはそれらの間を変動しているのかどうか)を確かめることができるようになるまで。STAP細胞由来Fgf4誘導幹細胞に関して言えば、それは胚およびまた胎盤組織の両方に寄与することができるものであるが、我々の阻害剤処理と組み合わせた試験管内転換研究は、Fgf4誘導幹細胞の双方向能力が培養皿の中にES様および栄養膜幹細胞様細胞の個別の細胞集団がコンタミしているようではないことを明らかに示している。総じて、我々の研究は、STAPを基盤とした転換が体細胞をリプログラムして、多能性だけでなく栄養膜分化能をも獲得させることを示している。
- 2019/05/14(火) 08:58:48|
- レター論文
-
-
| コメント:0
(英文)
Bidirectional developmental potential in reprogrammed cells with acquired pluripotency
Haruko Obokata,1, 2, 3,
Yoshiki Sasai,4,
Hitoshi Niwa,5,
Mitsutaka Kadota,6,
Munazah Andrabi,6,
Nozomu Takata,4,
Mikiko Tokoro,2,
Yukari Terashita,1, 2,
Shigenobu Yonemura,7,
Charles A. Vacanti3,
& Teruhiko Wakayama2, 8,
多能性獲得リプログラム細胞の双方向発生能力
Haruko Obokata
Yoshiki Sasai
Hitoshi Niwa
Mitsutaka Kadota
Munazah Andrabi
Nozomu Takata
Mikiko Tokoro
Yukari Terashita
Shigenobu Yonemura
Charles A. Vacanti
& Teruhiko Wakayama
(英文)
Journal name:
Nature
Volume:
505,
Pages:
676–680
Date published:
(30 January 2014)
DOI:
doi:10.1038/nature12969
雑誌名
ネイチャー
巻:
505,
頁
676–680
公開日
(2014/1/30)
DOI:
doi:10.1038/nature12969
(英文)
Received
10-Mar-13
Accepted
20-Dec-13
Published online
29-Jan-14
Retraction (July, 2014)
受領
2013/3/10
受諾
2013/12/20
オンライン公開
2014/1/29
取下げ(2014年7月)
(英文)
Abstract
We recently discovered an unexpected phenomenon of somatic cell reprogramming into pluripotent cells by exposure to sublethal stimuli, which we call stimulus-triggered acquisition of pluripotency (STAP)1. This reprogramming does not require nuclear transfer2, 3 or genetic manipulation4. Here we report that reprogrammed STAP cells, unlike embryonic stem (ES) cells, can contribute to both embryonic and placental tissues, as seen in a blastocyst injection assay. Mouse STAP cells lose the ability to contribute to the placenta as well as trophoblast marker expression on converting into ES-like stem cells by treatment with adrenocorticotropic hormone (ACTH) and leukaemia inhibitory factor (LIF).
In contrast, when cultured with Fgf4, STAP cells give rise to proliferative stem cells with enhanced trophoblastic characteristics. Notably, unlike conventional trophoblast stem cells, the Fgf4-induced stem cells from STAP cells contribute to both embryonic and placental tissues in vivo and transform into ES-like cells when cultured with LIF-containing medium. Taken together, the developmental potential of STAP cells, shown by chimaera formation and in vitro cell conversion, indicates that they represent a unique state of pluripotency.
概要
我々は最近、亜致死刺激に曝すことによって多能性細胞へリプログラミングされる体細胞の予期しない現象を発見した。我々はそれを刺激惹起性多能性獲得(STAP)[1]と呼ぶ。この再プログラミングは核移植[2][3]または遺伝子操作[4]を必要としない。ここに我々は、再プログラミングされたSTAP細胞が、胚盤胞注入実験に見られるように、胚性幹細胞(ES)とは異なり、胚および胎盤組織の両方に寄与し得ることを報告する。マウスSTAP細胞は、副腎皮質刺激ホルモン(ACTH)、および白血病抑制因子(LIF)で処理することにより、ES様幹細胞に変換する際に、胎盤に貢献する能力ならびに栄養膜マーカー発現を失う。
対して、Fgf4で培養すると、STAP細胞は、強化された栄養膜特性を伴った増殖幹細胞になる。特に、従来の栄養膜幹細胞とは異なり、STAP細胞からFGF4によって誘導された幹細胞は体内で胚および胎盤組織の両方に寄与し、LIF含有培地で培養するとES様細胞に形質転換する。まとめると、キメラの形成と試験管内細胞変換によって示されたSTAP細胞の発生能は、それらが多能性の特殊な状態を表すことを示している。
(英文)
We recently discovered an intriguing phenomenon of cellular fate conversion: somatic cells regain pluripotency after experiencing sublethal stimuli such as a low-pH exposure. When splenic CD45+ lymphocytes are exposed to pH 5.7 for 30 min and subsequently cultured in the presence of LIF, a substantial portion of surviving cells start to express the pluripotent cell marker Oct4 (also called Pou5f1) at day 2. By day 7, pluripotent cell clusters form with a bona fide pluripotency marker profile and acquire the competence for three-germ-layer differentiation as shown by teratoma formation. These STAP cells can also efficiently contribute to chimaeric mice and undergo germline transmission using a blastocyst injection assay.
Although these characteristics resemble those of ES cells, STAP cells seem to differ from ES cells in their limited capacity for self-renewal (typically, for only a few passages) and in their vulnerability to dissociation. However, when cultured in the presence of ACTH and LIF for 7 days, STAP cells, at a moderate frequency, further convert into pluripotent ‘stem’ cells that robustly proliferate (STAP stem cells).
我々は最近細胞運命変換の興味深い現象を発見した。即ち、体細胞が低pHにさらすなどの亜致死刺激を経験した後に多能性を取り戻すというものである。脾臓CD45+リンパ球がpH5.7に30分さらされ、その後LIFの存在下で培養されたとき、生存細胞のかなりの割合が2日目に多能性細胞マーカーOct4(Pou5f1とも呼ばれる)を発現し始める。7日目までに多能性細胞塊が真正な多能性マーカープロファイルをともなってでき、同時に奇形腫形成によって示されるような三胚葉分化能を獲得する。これらのSTAP細胞はまた、胚盤胞注入実験を用いて、確実にキメラマウスに寄与でき、生殖系列伝達を確認できる。
これらの特性は、ES細胞のものに似ているが、STAP細胞は、自己増殖能が弱い点と(典型的にはわずか数継代)、解離脆弱性において、ES細胞と異なっているように見える。 しかし、7日間ACTHおよびLIFの存在下で培養されると、STAP細胞は、かなりの頻度で、さらに、確実に増殖する多能性幹細胞(STAP幹細胞)になる。
(英文)
Here we have investigated the unique nature of STAP cells, focusing on their differentiation potential into the two major categories (embryonic and placental lineages) of cells in the blastocyst. We became particularly interested in this question after a blastocyst injection assay revealed an unexpected finding. In general, progeny of injected ES cells are found in the embryonic portion of the chimaera, but rarely in the placental portion (Fig. 1a; shown with Rosa26-GFP). Surprisingly, injected STAP cells contributed not only to the embryo but also to the placenta and fetal membranes (Fig. 1b and Extended Data Fig. 1a–c) in 60% of the chimaeric embryos (Fig. 1c).
ここでは、胚盤胞内細胞の二つの主要なカテゴリー(胚と胎盤関連)への分化能に焦点を当て、STAP細胞の特殊な性質を調査した。我々は胚盤胞注入実験が意外な発見を明らかにした後、この疑問に特に興味を持つようになった。一般的には、注入されたES細胞の所産はキメラの胚部分に見いだされるが、めったに胎盤部分には見いだされ無い(図1A、Rosa26-GFPで示されている)。驚くべきことに、STAP細胞は、キメラ胚の60%において(図1c)、胚のみならず胎盤と卵黄嚢(図1b及び拡張データ図1a-c)に貢献した。
(英文)
Figure 1: STAP cells contribute to both embryonic and placental tissues in vivo.
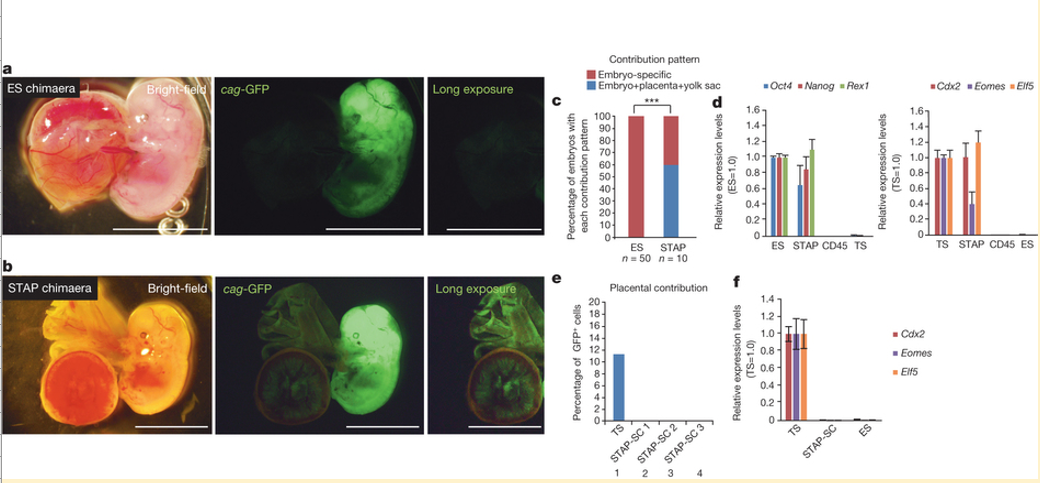
a, b, E12.5 embryos from blastocysts injected with ES cells (a) and STAP cells (b). Both cells are genetically labelled with GFP driven by a constitutive promoter. Progeny of STAP cells also contributed to placental tissues and fetal membranes (b), whereas ES-cell-derived cells were not found in these tissues (a). Scale bar, 5.0 mm. c, Percentages of fetuses in which injected cells contributed only to the embryonic portion (red) or also to placental and yolk sac tissues (blue). ***P < 0.001 with Fisher’s exact test. d, qPCR analysis of FACS-sorted Oct4-GFP-strong STAP cells for pluripotent marker genes (left) and trophoblast marker genes (right). Values are shown as ratio to the expression level in ES cells. Error bars represent s.d.
e, Contribution to placental tissues. Unlike parental STAP cells and trophoblast stem (TS) cells, STAP stem cells (STAP-SCs) did not retain the ability for placental contributions. Three independent lines were tested and all showed substantial contributions to the embryonic portions. f, qPCR analysis of trophoblast marker gene expression in STAP stem cells. Error bars represent s.d.
図1:STAP細胞は、体内実験で、胚および胎盤の両方の組織に寄与する。
a, b,ES細胞(a)およびSTAP細胞(b)を注入後の胚盤胞からのE12.5胚。どちらの細胞も、遺伝子操作により、構成的プロモーターで駆動されるGFP標識されている。 ES細胞由来の細胞はこれらの組織で検出されなかったのに対し(a)、STAP細胞の所産はまた、胎盤組織および胎児の膜(b)に貢献した。スケールバー、5.0 mm。 c, 注入された細胞が胚部分(赤)にのみ貢献したか、或いは胎盤と卵黄嚢組織(青)にも貢献しているかの胎児のパーセンテージ 。*** フィッシャーの正確確率検定P値<0.001。 d, 多能性マーカー遺伝子(左)と栄養膜マーカー遺伝子(右)に関する、FACSソートしたOct4-GFP-強発現STAP細胞の定量PCR分析。値はES細胞における発現レベルに対する比として示している。エラーバーは±標準偏差を表す。<訳注 s.d.=standard deviation>
e, 胎盤組織への貢献。親のSTAP細胞と栄養膜幹(TS)細胞とは異なり、STAP幹細胞(STAP-SCs)は、胎盤貢献能を保持していなかった。三つの独立した系統を試験し、すべて胚の部分には多大な貢献を示した。 f,STAP幹細胞の栄養膜マーカー遺伝子発現の定量PCR分析。エラーバーは±標準偏差を表す。
(英文)
In quantitative polymerase chain reaction (qPCR) analysis, STAP cells (sorted for strong Oct4-GFP signals) expressed not only pluripotency marker genes but also trophoblast marker genes such as Cdx2 (Fig. 1d and Supplementary Table 1 for primers), unlike ES cells. Therefore, the blastocyst injection result is not easily explained by the idea that STAP cells are composed of a simple mixture of pluripotent cells (Oct4+Cdx2-) and trophoblast-stem-like cells (Oct4-Cdx2+).
In contrast to STAP cells, STAP stem cells did not show the ability to contribute to placental tissues (Fig. 1e, lanes 2–4), indicating that the derivation of STAP stem cells from STAP cells involves the loss of competence to differentiate into placental lineages. Consistent with this idea, STAP stem cells show little expression of trophoblast marker genes (Fig. 1f).
定量ポリメラーゼ連鎖反応(qPCR)分析では、(Oct4-GFPシグナル強発現選別された)STAP細胞はES細胞とは異なり、多能性マーカー遺伝子だけでなく、Cdx2のような栄養膜マーカー遺伝子(図1dおよびプライマーのための補足表1)を発現した。したがって、胚盤胞注入結果は、STAP細胞が、多能性細胞(Oct4+ Cdx2-)と栄養膜幹細胞様細胞(Oct4-Cdx2+)との単純な混合で構成されているという考えでは容易に説明されない
STAP細胞とは対照的に、STAP幹細胞は、胎盤組織に貢献する能力を示さなかった(図1e 2-4レーン)。このことはSTAP細胞からSTAP幹細胞への誘導は、胎盤関連への分化能の喪失を伴うことを示している。その証拠にSTAP幹細胞は栄養膜マーカー遺伝子をほとんど発現しない(図1f)。
(英文)
We next examined whether an alteration in culture conditions could induce in vitro conversion of STAP cells into cells similar to trophoblast stem cells, which can be derived from blastocysts during prolonged adhesion culture in the presence of Fgf4. When we cultured STAP cell clusters under similar conditions (Fig. 2a; one cluster per well in a 96-well plate), flat cell colonies grew out by days 7–10 (Fig. 2b, left; typically in ~30% of wells). The Fgf4-induced cells strongly expressed the trophoblast marker proteins integrin α7 (Itga7) and eomesodermin (Eomes) (Fig. 2c, d) and marker genes (for example, Cdx2; Fig. 2e).
我々は、次に、培養条件の変更で、STAP細胞から栄養膜幹細胞に似た細胞への試験管内変換を誘導できるかどうかを調べた。栄養膜幹細胞はFgf4の存在下で長期接着培養中の胚盤胞から誘導することができる。同様の条件下でSTAP細胞塊を培養した場合(図2a、96ウェルプレートにおけるウェルあたり1クラスタ)、7日から10日までの間に扁平な細胞コロニーが成長してきた(図2b、左:典型的には、ウェルの30%内 )。Fgf4誘導細胞は栄養膜マーカー蛋白質であるインテグリンα7(Itga7)とエオメソダミン(Eomes)(図2c,d)及びマーカー遺伝子(たとえばCdx2;図2e)を強く発現した。
- 2019/05/14(火) 08:41:25|
- レター論文
-
-
| コメント:0
(英文)
STAP stem-cell conversion culture
1. To establish STAP stem-cell lines, STAP cell clusters were transferred to ACTH-containing medium on MEF feeder cells (several clusters, up to a dozen clusters, per well of 96-well plates).
IMPORTANT
(i) ACTH (1-24) is available from American Peptide and other companies. We used ACTH synthesized by Kurabo on consignment. The composition of this medium is GMEM, 15% Knockout Serum ReplacementTM (KSR, Invitrogen), 1 × non-essential amino acids (NEAA), 1 × Sodium Pyruvate, 10-4M 2-mercaptoethanol, 1000 U/ml LIF, and 10 µM ACTH . The STAP cell cluster was isolated, dissected into small pieces as in the case of injection into blastocysts as shown in Figure 4a (Obokata et al. Nature, 2014a), and seeded on mouse embryonic fibroblast feeder cells in the ACTH medium.
(ii) ACTH-containing medium was purchased from DS Pharma Biomedical (Osaka, Japan) as StemMedium@.
STAP幹細胞変換培養
1. STAP幹細胞株を確立するために、 STAP細胞クラスターはMEFフィーダー細胞上のACTH含有培地に移される。(96ウェルプレートの1ウェルあたり1ダースまでの複数のクラスタ)
[重要]
(i)ACTH(1-24)は、アメリカンペプチド社および他の会社から入手可能である。我々は委託契約でクラボウによって合成されたACTHを使用していた。この培地の組成は、GMEM、15%ノックアウト血清代替TM(KSR、Invitrogen社)、1×非必須アミノ酸(NEAA)、1×ピルビン酸ナトリウム、10 -4 M2-メルカプトエタノール、1000U / mlのLIF、および10μM ACTHである (Ogawa et al, Genes Cells, 2004)。 STAP細胞のクラスターは図4a (Obokata et al. Nature, 2014a)にしめされた胚盤胞注入の場合のように単離され小片に切開され、ACTH培地中のマウス胚線維芽細胞フィーダー細胞上に播種された。
(ii)ACTH含有培地はステムミージャムアドレスのDSファーマバイオメディカル社(大阪、日本)から購入されている。
(英文)
2. After 4–7 days of culture, the cells were subjected to a first passage using a conventional trypsin method, and the suspended cells were plated in ESC maintenance medium containing 20% FBS.
IMPORTANT
(i) ESC maintenance medium consists of KnockoutTM DMEM (Life Technologies), 20% FBS, 1 × NEAA, 1 × Glutamine, 1 × Nucleosides, 10-4M 2-mercaptoethanol, and 1000 U/ml LIF.
(ii) FBS lots should be confirmed for suitability for use in the culture of mouse ES cells.
(iii) We have established multiple STAP stem cell lines from STAP cells derived from CD45+ haematopoietic cells. Of eight clones examined, none contained the rearranged TCR allele, suggesting the possibility of negative cell-type-dependent bias (including maturation of the cell of origin) for STAP cells to give rise to STAP stem cells in the conversion process. This may be relevant to the fact that STAP cell conversion was less efficient when non-neonatal cells were used as somatic cells of origin in the current protocol.
2. 培養の4から7日後に、細胞は従来のトリプシン法を使って第一段階に供され、懸濁した細胞を、20%FBS<ウシ胎児血清 fetal bovine serum>を含むES細胞維持培地に蒔種された。
[重要]
(i)ES細胞維持培地は KnockoutTM DMEM<イーグル最小必須培地>(Life Technologies社)、20%FBS、1×NEAA、1×グルタミン、1×ヌクレオシド、10 -4M 2-メルカプトエタノール及び1000 U/mlのLIFから構成されている。
(ii)のFBSロットは、マウスES細胞の培養に使用するための適合性が確認されるべきである。
(ⅲ)我々はCD45陽性造血細胞に由来するSTAP細胞から複数のSTAP幹細胞株を確立している。調べた8個のクローンのうちのいずれも再構成されたTCR対立遺伝子を含んでいなかった。このことはSTAP細胞が変換プロセスでSTAP幹細胞になるための、(もとの細胞の突然変異含む)陰性の細胞型依存バイアスの可能性を示唆している。これは現在のプロトコルにおいて、非新生細胞をもとの体細胞として使用したときに、STAP細胞の変換がそれほど効率的でなかったという事実に関係しているかも知れない。
(英文)
3. Subsequent passaging was performed at a split ratio of 1:10 every second day until reaching subconfluency. We tested the following three different genetic backgrounds of mice for STAP stem-cell establishment from STAP cell clusters, and observed reproducible establishment: C57BL/6 carrying Oct4-gfp (29 of 29), 129/Sv carrying Rosa26-gfp (2 of 2), and 129/Sv × C57BL/6 carrying cag-gfp (12 of 16). STAP stem cells with all these genetic backgrounds showed chimaera-forming activity.
3. その後の継代はサブコンフルエント(シャーレ一杯になる前)に達するまでの二日置きに、1対10の分割比で行なった。我々はSTAP細胞クラスターからSTAP幹細胞を樹立するために、マウスの以下の3つの異なる遺伝的背景をテストし、再現性の確立を観察した。Oct4-GFP(29の29)を持つC57BL/6、Rosa26-GFP(2の2)をもつ129/ SV、および129/ CAG-GFP(16の12)をもつSV×C57BL/6。すべてのこれらの遺伝的背景を持つSTAP幹細胞はキメラ形成活性を示した。
(英文)
FI stem cell conversion culture
1. STAP cell clusters were transferred to Fgf4-containing trophoblast stem-cell medium (Tanaka et al, Science, 1998) on MEF feeder cells in 96-well plates (Obokata, Nature, 2014b).
IMPORTANT
(i) TS medium consists of RPMI 1640 with 20% FBS, 1 mM Sodium Pyruvate, 100 µM 2-mercaptoethanol, 2 mM L-glutamine, 25 ng/ml of recombinant FGF4, and 1 µg/ml of heparin.
(ii) Different lots of FBS may results in significant differences in the behavior of cultured cells.
2. In most cases (40 of 50 experiments), colonies grew in 10–50% of wells in 96-well plates. In a minority of cases (10 of 50 experiments), no colony growth was observed and/or only fibroblast-like cells appeared.
IMPORTANT
(i) The cells in proliferative colonies also appear similar to fibroblasts, but gradually change morphology, coming to resemble epithelial cells.
FI幹細胞変換培養
1. STAP細胞クラスターがFGF4を含む96ウェルプレート中のMEFフィーダー細胞上のTS細胞<栄養芽層幹細胞>培地 (Tanaka et al, Science, 1998)に移される (Obokata, Nature, 2014b)。
[重要]
(i)TS培地は20%のFBS<ウシ胎児血清>を含むRPMI1640<ロズウェルパーク記念研究所培地 Roswell Park Memorial Institute medium>、1mMのピルビン酸ナトリウム、100μMの2-メルカプトエタノール、2mMのL-グルタミン、組換えFGF4の25 ng / ml及びヘパリンの1μg/mlで構成されている。
(ii)FBSのロットの違いは培養細胞の挙動に重大な差異をもたらしうる。
2. ほとんどの場合(50回のうちの40回の実験)、コロニーは96ウェルプレートのウェルの10から50%までに成長した。少数の例(50のうちの10回の実験)では、全くコロニー増殖が観察されずに、かつ/または、わずかの線維芽細胞様細胞が出現した。
重要
(i)増殖コロニー内の細胞はまた線維芽細胞のように現れるが、徐々に形態を変更して上皮細胞に似てくる。
(英文)
3. The cells were subjected to the first passage during days 7–10 using a conventional trypsin method. Subsequent passages were performed at a split ratio of 1:4 every third day before they reached subconfluency.
IMPORTANT
The cells must not be dissociated completely. Partial dissociation is optimal to maintain viability and self-renewal, as seen in the case of embryo-derived trophoblast stem cells.
3. 細胞を従来のトリプシン法を用いて7-10日間の最初の継代に供した。それらがサブコンフルエントに達する前に3日おきに4対1の分割比でその後の継代を行った。
[重要]
(i) 細胞は完全に解離されてはいけない。胚由来のトロホブラスト幹細胞の場合に見られるように、部分的な解離が生存能力および自己再生を維持するのに最適である。
(英文)
References
Obokata, H. et al.Obokata. Stimulus-triggered fate conversion of somatic cells into pluripotency, Nature, 505, 641-647 (2014a)
Obokata, H. et al. Bidirectional developmental potential in reprogrammed cells with acquired pluripotency. 676–680 (2014b)
Ohbo, K. et al. Identification and characterization of stem cells in prepubertal spermatogenesis in mice small star, filled. Dev. Biol. 258, 209–225 (2003)
Yoshimizu, T. et al. Germline-specific expression of the Oct4/green fluorescent protein (GFP) transgene in mice. Dev. Growth. Differ. 6, 675-684 (1999)
Ogawa, K., Matsui, H., Ohtsuka, S. & Niwa, H. A novel mechanism for regulating clonal propagation of mouse ES cells. Genes Cells 9, 471–477 (2004)
Tanaka, S., Kunath, T., Hadjantonakis, A. K., Nagy, A. & Rossant, J. Promotion of trophoblast stem cell proliferation by FGF4. Science 282, 2072–2075 (1998)
<参照>
Obokata, H. et al.Obokata. 『体細胞の多能性への刺激惹起性運命変換』Nature, 505, 641-647 (2014a)
Obokata, H. et al. 『取得多能性を持つ再プログラム細胞における双方向への発生能力』 676–680 (2014b)
Ohbo, K. et al. 『マウスの小さな星の中に満たされた思春期前の精子形成における幹細胞の同定と特徴づけ』 Dev. Biol. 258, 209–225 (2003)
Yoshimizu, T. et al. 『マウスのOct4/緑色蛍光タンパク質(GFP)導入遺伝子の生殖細胞特異的発現』 Dev. Growth. Differ. 6, 675-684 (1999)
Ogawa, K., Matsui, H., Ohtsuka, S. & Niwa, H. 『マウスES細胞のクローン増殖を調節するための新しいメカニズム』 Genes Cells 9, 471–477 (2004)
Tanaka, S., Kunath, T., Hadjantonakis, A. K., Nagy, A. & Rossant, J. 『FGF4<線維芽細胞増殖因子>によるTS細胞増殖の推進』 Science 282, 2072–2075 (1998)
(英文)
Associated Publications
This protocol is related to the following articles:
Stimulus-triggered fate conversion of somatic cells into pluripotency
Haruko Obokata, Teruhiko Wakayama, Yoshiki Sasai, Koji Kojima, Martin P. Vacanti, Hitoshi Niwa, Masayuki Yamato, and Charles A. Vacanti
Journal title
Nature
Vol.
505
Issue
-7485
Pages
641 - 647
Publication date
29/01/2014
DOI:
doi:10.1038/nature12968
<関連著作物>
このプロトコルは下記論文に関連している。
『体細胞の多能性への刺激惹起性運命変換』
Haruko Obokata, Teruhiko Wakayama, Yoshiki Sasai, Koji Kojima, Martin P. Vacanti, Hitoshi Niwa, Masayuki Yamato, and Charles A. Vacanti
Journal title
Nature
Vol.
505
Issue
-7485
Pages
641 - 647
Publication date
29/01/2014
DOI:
doi:10.1038/nature12969
(英文)
Bidirectional developmental potential in reprogrammed cells with acquired pluripotency
Haruko Obokata, Yoshiki Sasai, Hitoshi Niwa, Mitsutaka Kadota, Munazah Andrabi, Nozomu Takata, Mikiko Tokoro, Yukari Terashita, Shigenobu Yonemura, Charles A. Vacanti, and Teruhiko Wakayama
Journal title
Nature
Vol.
505
Issue
-7485
Pages
676 - 680
Publication date
29/01/2014
DOI:
doi:10.1038/nature12969
『取得多能性を持つ再プログラム細胞における双方向への発生能力』
Haruko Obokata, Yoshiki Sasai, Hitoshi Niwa, Mitsutaka Kadota, Munazah Andrabi, Nozomu Takata, Mikiko Tokoro, Yukari Terashita, Shigenobu Yonemura, Charles A. Vacanti, and Teruhiko Wakayama
Journal title
Nature
Vol.
506
Issue
-7484
Pages
677 - 680
Publication date
29/01/2015
DOI:
doi:10.1038/nature12970
(英文)
Author Information
Affiliations
1. Laboratory for Cellular Reprogramming, RIKEN CDB
Haruko Obokata
2. Laboratory for Organogenesis and Neurogenesis, RIKEN CDB
Yoshiki Sasai
3. Laboratory for Pluripotent Stem Cell Studies, RIKEN CDB
Hitoshi Niwa
Competing financial interests
The authors declare no competing financial interests.
Corresponding author
Correspondence to:
Hitoshi Niwa (niwa@cdb.riken.jp)
<著者紹介>
[所属]
1.細胞リプログラミング研究室、理研CDB
小保方晴子
2.器官発生・神経発生研究室、理研CDB
笹井芳樹
3.多能性幹細胞解明研究室、理研CDB
丹羽仁
[金銭的利益相反]
著者等は金銭的利益相反のないことを宣誓します。
[責任著者]
連絡はこちらへ
Hitoshi Niwa (niwa@cdb.riken.jp)
- 2019/05/14(火) 08:24:42|
- プロトコル
-
-
| コメント:0
(英文)
Essential technical tips for STAP cell conversion culture from somatic cells
Haruko Obokata,
Yoshiki Sasai
& Hitoshi Niwa
STAP Group RIKEN CDB
Journal name:
Protocol Exchange
Year published:
-2014
DOI:
doi:10.1038/protex.2014.008
Published online
5-Mar-14
体細胞からのSTAP細胞変換培養に欠かせない技術上の秘訣
Haruko Obokata,
Yoshiki Sasai
& Hitoshi Niwa
STAP Group RIKEN CDB
Journal name:
Protocol Exchange
Year published:
-2014
DOI:
doi:10.1038/protex.2014.008
Published online
5-Mar-14
(英文)
THE AUTHORS ARE RETRACTING THIS PROTOCOL EXCHANGE PROTOCOL BECAUSE THE PAPERS UPON WHICH IT IS BASED HAVE BEEN RETRACTED.
本論文が取り下げられていますので、作者らはこのプロトコルイクスチェンジのプロトコルを取り下げています。
(英文)
Abstract
Stimulus-triggered acquisition of pluripotency (STAP) is a cellular reprogramming phenomenon that was recently reported in two papers (Obokata, Nature, 2014a,b). In this reprogramming process, upon strong external stimuli, neonatal somatic cells are converted into cells that express pluripotency-related genes, such as Oct3/4, and acquire the ability to differentiate into derivatives of all three germ layers in vitro and in vivo. These cells, termed STAP cells, can contribute to chimeric fetuses after blastocyst injection. Moreover, in the blastocyst injection assay, injected STAP cells are also found in extra-embryonic tissues, such as placenta.
概要
刺激惹起性多能性獲得(STAP)は最近2つの論文で報告された細胞の再プログラミング現象である (Obokata, Nature, 2014a,b)。この再プログラミングプロセスでは、強力な外部刺激に応じて、新生児の体細胞が、Oct3/4のような多能性関連遺伝子を発現し、体外体内および体内で、三胚葉すべての派生物に分化する能力を獲得した細胞に変換される。、STAP細胞と定義されたこれらの細胞は胚盤胞注入後にキメラ胎児に寄与することができる。それにとどまらず、胚盤胞注入試験において、注入されSTAP細胞はまた胎盤などの胚の外部組織においても見出される。
(英文)
STAP cells derived from neonatal somatic cells are thus fully reprogrammed to as state of pluripotency. In the conditions for the establishment of STAP cells, their proliferative capacity is quite limited, distinct from that of embryonic stem cells (ESCs). STAP cells can be further converted into two types of proliferative cell lines: STAP stem cells and FGF4-induced stem cells (FI stem cells). STAP stem cells, which are converted from STAP cells in ACTH-containing medium (see Procedure), lose the ability to contribute to extra-embryonic tissues. FI stem cells, which are generated from STAP cells in FGF4-containing medium, in contrast retain the capacity to contribute to both embryonic and extra-embryonic lineages in blastocyst injection assay, although their embryonic contribution is relatively low.
新生児の体細胞に由来するSTAP細胞はこのように多能性状態に完全に再プログラムされている。 STAP細胞の樹立のための条件下では、それらの増殖能力は、胚性幹細胞(ESCs)のものとは異なり、非常に制限されている。 STAP細胞はさらに二つのタイプ増殖性細胞株に変換することができる。STAP幹細胞およびFGF4誘発性幹細胞(FI幹細胞)である。ACTH含有培地の中で(手順参照)、STAP細胞から変換されたSTAP幹細胞は、胚の外部組織に貢献する能力を失う。FGF4含有培地中の中でSTAP細胞から生成されたFI幹細胞は、それらの胚への寄与は比較的低いが、胚盤胞注入検査で、胚および胚の外部系統の両方に貢献する能力を保持する。
(英文)
The STAP phenomenon induced by external stimuli, thus potentially sheds new light on our understanding of pluripotency and differentiation in mammalian cells. This unforeseen phenomenon can be triggered in neonatal hematopoietic cells, for instance, by transient exposure to low-pH solution. Despite its seeming simplicity, this procedure requires special care in cell handling and culture conditions, as well as in the choice of the starting cell population. The delivery of the optimal level of sublethal stress to cells is essential to the process of STAP cell induction. From our experience, STAP conversion is reproducibly seen with culture conditions in which most cells survive for one day after low-pH treatment, and in which up to 80% of the initial cell number subsequently die at around days 2–3. Control of the pH of the solution is not the only key factor; the delayed onset of sublethal stress is also critically important.
外部刺激によって誘導されるSTAP現象は潜在的に哺乳動物細胞における多能性および分化に関する我々の理解に新たな光を投げかけるものである。この予期しない現象は例えば低pH溶液に過渡の曝すことによって新生児の造血細胞で惹起されうるものである。その見かけの単純さにかかわらず、この手順では細胞の取り扱いや培養条件のみならず、最初の細胞集団の選択にも特別な注意が必要である。細胞に最適レベルの亜致死刺激を与えることがSTAP細胞誘導の過程に不可欠である。我々の経験では、STAP変換はほとんどの細胞が低pH処理後一日生存し、その後初期細胞数の80%までが2から3日位で死亡するような培養条件で再現的に見られる。溶液のpHの調整だけが重要な要因ではなく、亜致死ストレスの遅発性も非常に重要である。
(英文)
This biological context can also be affected by many other factors. For example, somatic cell preparation and cell handling before and after the exposure to stress must be done with care, as additional damage to the cells may alter the level of stress, causing excessive cell death or insufficient triggering. The types of cells used for STAP conversion are also critical, and the use of cells from other sources (e.g., the use of cultured fibroblasts after passaging) may also result a failure to achieve STAP conversion. We have reproducibly observed STAP cell conversion when proper procedures are followed in the correct sequence.
この生物学的状況はまた他の多くの要因によっても影響され得る。例えばストレスにさらす前後の体細胞の準備と取り扱いは、過剰な細胞死か、不十分な惹起性が引き起こされた場合に、細胞への追加ダメージがストレスレベルを変更することができるように、注意深く行なわれなければならない。STAP変換に使用される細胞のタイプも重要であり、他のソースからの細胞の使用(例えば、継代後の培養線維芽細胞の使用)もSTAP変換を達成するために障害をもたらし得る。適切な手順が正しい順序で行なわれている場合に我々はSTAP細胞の変換を再現的に観察している。
(英文)
To facilitate the broad testing and use of this technique, we are now preparing a full protocol article with step-by-step instructions. However, as the preparation, submission and publication of a full manuscript takes a significant amount of time, we would like to share a number of technical tips for STAP cell conversion culture (and related experiments) in this Protocol Exchange. We hope that these technical tips may answer many questions frequently asked about the experimental details.
この技術の広範なテストと使用を容易にするために我々は今、ステップバイステップ手順の完全なプロトコル論文を準備中です。しかしながら、完全な原稿の作成、提出、公表にはかなりの時間がかかるので、我々は、このProtocol ExchangeでSTAP細胞の変換培養(および関連の実験)のための多くの技術的な秘訣を共有したいと思います。我々はこれらの技術的な秘訣が実験の詳細についてしばしば問われる多くの質問に答えることになることを願っています。
(英文)
Procedure
Tissue collection and low-pH treatment
1. To isolate CD45+ haematopoietic cells, spleens were excised from 1-week-old Oct4-gfp mice (unless specified otherwise), minced by scissors, and mechanically dissociated using a Pasteur pipette.
IMPORTANT
(i) Adherent cells should be dissociated into single cells, either mechanically or enzymatically (by trypsin or collagenase). For the tissues described in Fig. 3a (Obokata et al. Nature, 2014a), muscle, adipose tissue and fibroblasts were enzymatically dissociated, whereas others were mechanically dissociated.
(ii) Primary cells should be used. We have found that it is difficult to reprogram mouse embryonic fibroblasts (MEF) that have been expanded in vitro, while fresh MEF are competent.
(iii) For the experiments reported, we used a Oct-3/4-EGFP transgenic mouse line (Ohbo et al, Dev Biol, 2003; Yoshimizu et al, Dev Growth Differ, 1999), which is maintained by the RIKEN Bioresource Center as GOF18-GFP line11 transgenic mouse (B6;B6D2-Tg(GOF18/EGFP)11/Rbrc). Homozygotes of the transgene were used for the live imaging to obtain the enhanced signal.
(iv) Cells from mice older than one week showed very poor reprogramming efficiency under the current protocol. Cells from male animals showed higher efficiency than those from female.
<手順>
組織収集および低pH処理
1. CD45陽性造血細胞を単離するために、1週齢のOct4-GFPマウス(特に指定のない限り)から脾臓が摘出され、ハサミによりミンチされ、そして機械的にパスツールピペットを用いて分離される。
[重要]
(i) 接着細胞は機械的または酵素的に(トリプシンまたはコラゲナーゼにより)単一細胞に解離されなければならない。図3a (Obokata et al. Nature, 2014a)で説明されている組織のうち、他の細胞が機械的に解離させられたのに対して、筋肉、脂肪組織および線維芽細胞は酵素的に解離させられている。
(ii)一次細胞が使用されるべきである。我々は新鮮なマウス胚線維芽細胞(MEF)なら可能であるが、体外で増殖されたMEFを再プログラムすることは困難であることをすでに知っている。
(iii) 報告されている実験のために、我々は、理化学研究所バイオリソースセンターによってGOF18-GFP株11トランスジェニックマウス(B6;B6D2-Tg(GOF18/EGFP)11/Rbrc)として維持されている、Oct-3/4-EGFPトランスジェニックマウス株を使っている(Ohbo et al, Dev Biol, 2003; Yoshimizu et al, Dev Growth Differ, 1999)。導入遺伝子のホモ接合体は、強化された信号を得るため、ライブイメージング用に使用されている。
(iv)1週間以上経過したマウス由来の細胞は現在のプロトコルの下では非常に貧弱な再プログラミング効率を示した。雄の動物からの細胞は雌からのものよりも高い効率を示している。
(英文)
2. Dissociated spleen cells were suspended with PBS and strained through a cell strainer (BD Biosciences 352340).
3. After centrifuging at 1,000 rpm for 5 min, collected cells were re-suspended in DMEM medium and added to the same volume of lympholyte (Cedarlane), and then centrifuged at 1,000 g for 20 min.
IMPORTANT
(i) The purity of the starting cells is important for achieving STAP conversion. For lymphocytes, contamination with red blood cells may inhibit the reprogramming event. When using adherent cells, the presence of extracellular matrix may interfere with reprogramming.
(ii) Alternatively, red blood cells may be removed by suspension of the cell pellet in 1.8 ml of H2O (Sigma W3500). After 30 seconds, add 0.2 ml of 10× PBS (Gibco 70011-044), followed by 3 ml of 1× PBS (Gibco 10010-023), and strain the cell suspension through a cell strainer.
2. 解離脾臓細胞をPBS<リン酸緩衝生理食塩水 Phosphate buffered saline>で懸濁し、細胞濾過器(BD Biosciences社352340)で漉した。
3. 1000回転/分で5分間遠心分離した後、回収した細胞をDMEM培地<ダルベッコ変法イーグル培地 Dulbecco's modified Eagle medium>に再懸濁し、同じ体積のヒトリンパ球分離性溶液<lympholyt>(セダレーン社)を添加し、次いで20分間1000gで遠心分離した。
[重要]
(i)出発細胞の純度はSTAP変換を達成するために重要である。リンパ球にとって赤血球の混入が再プログラミング事象を阻害することがある。接着細胞を用いる場合には細胞外基質の存在が再プログラミングを妨害することがある。
(ii)代替手段として1.8 mlの水(シグマ社W3500)の中に細胞ペレットを懸濁することによって赤血球を除去してもよい。30秒後に、10倍濃度のPBS<リン酸緩衝生理食塩水>(ギブコ社70011-044)を0.2mlを、続いて1倍濃度のPBS(ギブコ社10010-023)3mlを加えて、細胞濾過器を通して細胞懸濁液を濾過する。
(英文)
4. The lymphocyte layer was isolated and stained with CD45 antibody (Abcam ab25603). CD45+ cells were sorted by FACS Aria (BD Biosciences).
IMPORTANT
(i) FACS sorting can be an important step for the confirmation of cell purity, but can affect both cell viability and reprogramming efficiency. Skipping this step may increase reprograming efficiency, although this may result in a reduction in confidence in cell identity.
5. After cell sorting, 1 × 106 CD45+ cells were treated with 500 µl of low-pH HBSS solution (titrated to pH 5.7 by HCl) for 25 min at 37°C, and then centrifuged at 1,000 rpm at room temperature for 5 min.
IMPORTANT
(i) The buffering action of HBSS is weak, so carry-over of the solution may affect pH. Please adjust pH to 5.7 in cell suspension by the following method. First, suspend the cell pellet with 494 µl of HBSS pre-chilled at 4°C, then add 6 µl of diluted HCl (10 µl of 35% HCl in 590 µl of HBSS) to adjust to a final pH of 5.7. Please confirm the final pH in a pilot experiment, and optimize the volume of HCl added, as necessary. Alternatively, suspend the cell pellet in HBSS-pH 5.4 pre-chilled at 4°C.
(ii) The HBSS we used is Ca2+/Mg2+ free (Gibco 14170-112).
(iii) Incubate the cells suspended in HBSS in a CO2 incubator.
(iv) Cell viability is a critical parameter in this step. Under optimal conditions, massive cell death is observed at two days after plating, as shown in Figure 1d (Obokata et al. Nature, 2014a).
(v) If you find massive cell death at one day after plating, it may be ameliorated by shortening the incubation period with low-pH HBSS solution to 15 min.
4. リンパ球層を単離し、CD45抗体(Abcam社のab25603)で染色した。 CD45+細胞をFACSアリア(BD Biosciences社)によって選別した。
[重要]
(i) FACSソーティングは細胞の純度を確保するための重要なステップであり得るが、細胞の生存率と初期化効率の両方に影響を与えうる。細胞出自の信頼性を減少させるかもしれないが、このステップをスキップすると初期化効率を高めることができる。
5. 細胞選別後、100万個のCD45陽性細胞を37℃で25分間、低pH(塩酸によりpHを5.7に滴定)のHBSS<ハンクス平衡塩溶液溶液>500μlで処理し、次いで室温で1000rpmで5分間遠心分離した。
[重要]
(i)HBSSの緩衝作用が弱いため、溶液のキャリーオーバーがpHに影響する可能性がある。以下の方法に従ってpHを5.7に調整してください。まず、事前に4℃に冷却されたHBSS494μlに細胞ペレットを懸濁し、次に、5.7の最終pHに調整するために希釈塩酸(HBSS590μlに35%塩酸を10μl)を6μl追加する。パイロット実験で最終pHを確認し、加える塩酸の量を必要に応じて最適化してください。そうでなければ、事前に4℃に冷却されたHBSS-pH5.4に細胞ペレットを懸濁してください。
(ii)我々の使用したHBSSはCa2+/Mg2+ free(ギブコ社14170-112)である。
(iii)炭酸ガス培養器内でHBSSに懸濁した細胞を培養してください。
(iv)細胞生存率はこの行程における重要なパラメータである。図1dに示すように、最適な条件下で、大量の細胞死がプレーティング後2日目に観察される (Obokata et al. Nature, 2014a)。
(v)プレーティング後一日目で大量の細胞死があった場合は、低pHのHBSS溶液での培養期間を15分に短縮することによって改善することができる。
(英文)
6. After the supernatant (low-pH solution) was removed, precipitated cells were re-suspended and plated onto non-adhesive culture plates (typically, 1×105 cells/ml) in DMEM/F12 medium supplemented with 1,000 U LIF (Sigma) and 2% B27 (Invitrogen).
IMPORTANT
(i) The use of non-adhesive culture plates is recommended, as the formation of cell clusters is an important step for reprogramming, and the adhesive surface may inhibit cell movement needed to form clusters.
(ii) Cell density is critical, and depends on cell viability. Density should be maintained at 1×105~1×106 cells per cm2 of culture surface.
(iii) B27 (Invitrogen 17504-044) may show variation between batches. Please check the quality by N2B27-2iLIF culture of ES cells.
6. 上澄み(低pH溶液)を除去した後、沈殿細胞を再懸濁し、千U LIF(シグマ社)と2%のB27(インビトロジェン社)を入れたDMEM / F12培地の中の非接着性培養プレートに播種する(通常、10万個細胞/ ml)。
[重要]
(i)非接着性培養プレートの使用が推奨される。なぜなら、細胞クラスターの形成は再プログラミングのための重要なステップであるが、接着性表面は、クラスタを形成するのに必要な細胞の移動を阻害しうるからである。
(ii)細胞密度は重要であり、それは細胞生存率に依存している。密度は培養表面1平方センチメートルあたり10万から100万個の細胞で維持されるべきである。
(iii)B27(インビトロジェン社17504-044)は商品バッチ間で違いのあることがある。 ES細胞のN2B27-2iLIF培養によって品質を確認してください。
(英文)
7. Cell cluster formation was more sensitive to plating cell density than to the percentage of Oct3/4-GFP+ cells. The number of surviving cells was sensitive to the age of donor mice, and was low under the treatment conditions described above when adult spleens were used.
IMPORTANT
(i) The donor mouse should be 1-week old or younger. Reprogramming efficiency is dramatically reduced using cells from older animals.
(ii) STAP cells are derived from clusters of multiple cells; they are not monoclonal.
8. The addition of LIF during days 2–7 was essential for generating Oct3/4-GFP+ STAP cell clusters on day 7, as shown in Extended Data Fig. 1f (Obokata et al. Nature, 2014a). Even in the absence of LIF, Oct3/4-GFP+ cells (most of which showed dim signals) appeared transiently in low-pH-treated CD45+ cells during days 2–5 of culture, but subsequently disappeared, suggesting that there is a LIF-independent early phase, whereas the subsequent phase is LIF-dependent.
IMPORTANT
(i) Since LIF is essential for the late step of reprogramming, the reprogramming event may depend on the genetic background. We mainly used 129, C57BL6, or their F1 strains, as all of these genetic backgrounds are associated with high responsiveness to LIF.
The GFP signal is weaker than that of ES cells carrying the same reporter because of the smaller cell volume of STAP cell than ES cell as found in Figure 1g (Obokata et al. Nature, 2014a).
7. 細胞のクラスター形成はOct3/4-GFP陽性細胞のパーセンテージに対してよりも、プレート移植されている細胞密度に対してより敏感であった。生存細胞数はドナーマウスの年齢に敏感であり、成長したマウス脾臓を用いた場合、上述した処理条件下で低かった。
[重要]
(i)ドナーマウスは1週齢もしくはもっと若くあるべきである。高齢の動物からの細胞を用いると再プログラミング効率は劇的に落ちる。
(ii)STAP細胞は複数の細胞のクラスタから誘導される。それらは単一種細胞ではない。
8. 2から7日目のにおけるのLIFの添加は、拡張データ図1fに示すように、7日目のOct3/4-GFP陽性STAP細胞クラスターを生成するために不可欠であった(Obokata et al. Nature, 2014a)。LIFの存在しない場合でも、Oct3/4-GFP陽性細胞(ほとんどが薄暗いシグナルを示したが)は培養2から5日の間に、低pH値で処理したCD45陽性細胞の中に一過的に現れたが、その後に姿を消した。LIF依存の次の段階に対してLIF非依存の初期段階のあることを示唆している。
[重要]
(i)再プログラミングの後期段階ではLIFは不可欠ですので、再プログラミング事象は遺伝的背景に依存しているのかも知れない。我々は主に129、C57BL6、またはそれらのF1株を使用したが、これらの遺伝的背景のすべてがLIFへの高い応答性に関連しているからである。
(ii) GFPシグナルは、同じレポーターを持っているES細胞のそれより弱いが、それは図1gに見られるように、STAP細胞の体積がES細胞よりも小さいためである(Obokata et al. Nature, 2014a)。
- 2019/05/14(火) 08:18:47|
- プロトコル
-
-
| コメント:0






